Introducción
La Inmunodeficiencia Común variable (IDCV) es una inmunodeficiencia primaria predominantemente de anticuerpos, en la cual la respuesta inmune humoral esta alterada 1,2. El espectro clínico de esta enfermedad va desde infecciones a repetición, con secuelas como la aparición de bronquiectasias, hasta el desarrollo de malignidad o autoinmunidad. A pesar de ser un trastorno genético, los adultos son los más afectados, por lo cual debe hacerse un esfuerzo en educar a la comunidad médica en general 2,3. A continuación presentamos el caso de un varón de 37 años con infecciones sinopulmonares a repetición y bronquiectasias generalizadas, en el cual se demostró una hipogamaglobulinemia severa con un cuadro compatible con Inmunodeficiencia Común variable.
Caso clínico
Hombre de 37 años de edad que se presentó al servicio de urgencias de un Hospital nivel III de la ciudad de Cali por dificultad respiratoria, fiebre y tos con expectoración verdosa de aproximadamente una semana de evolución, con empeoramiento de la disnea en las últimas 48 horas, hasta ser incapaz de realizar esfuerzos mínimos. Al ingreso hipotenso (78/36), taquicárdico (126 latidos/min), polipnéico (38 respiraciones/min) con Saturación de O2 del 76% (O2 ambiente), la auscultación pulmonar reveló múltiples sobre agregados y disminución global del murmullo vesicular. El paciente manifestó tener una inmunodeficiencia de anticuerpos y pocos minutos después del ingreso presenta falla ventilatoria, requiriendo intubación orotraqueal y soporte con ventilación mecánica y vasoactivos, se consideró choque séptico por lo cual se inició cubrimiento antibiótico con vancomicina y cefepime previa toma de hemocultivos.
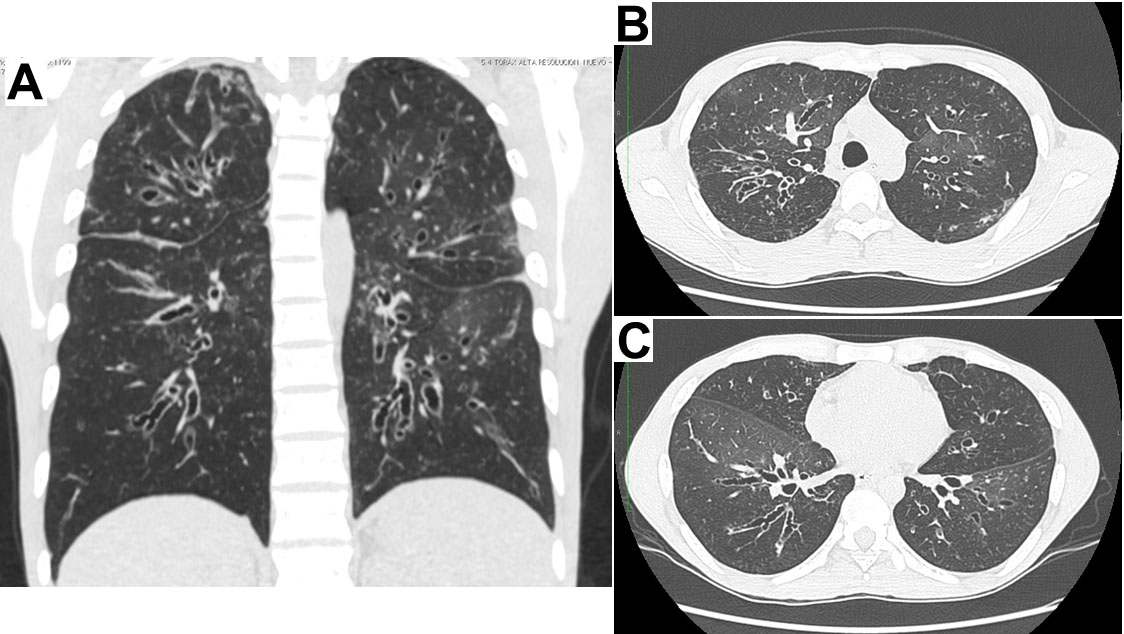
El paciente es originario y procedente de Cali (Valle), como antecedentes relevantes refiere neumonías, sinusitis y otitis a repetición desde los 18 años de edad, los episodios son innumerables (entre 6 y 10 por año) requiriendo largos cursos de antibióticos orales o endovenosos así como múltiples hospitalizaciones. Desde el 2002 se habían documentado bronquiectasias cilíndricas y quísticas en los cuatro cuadrantes (Fig. 1), se realizaron estudios para fibrosis quística y autoinmunidad los cuales fueron negativos. En 2006 recibió 6 meses de tratamiento acortado supervisado por tuberculosis pulmonar diagnosticada por cultivo de esputo positivo a las 12 semanas (baciloscopias seriadas negativas) con curación microbiológica. Había consultado en incontables oportunidades por diferentes especialidades y subespecialidades de medicina interna y a pesar de tener documentados títulos muy bajos de inmunoglobulinas séricas totales (al menos 5 años antes de esta hospitalización) no recibía terapia de reemplazo por razones poco claras, se le realizaron pruebas de anticuerpos para VIH en más de 7 oportunidades con resultados negativos.
Figura 1.
Escanografía de Tórax de alta resolución en la cual se evidencian bronquiectasias generalizadas en los cuatro cuadrantes en A y en los cortes transversales a nivel del cayado aórtico en B y a nivel del ventrículo izquierdo en C
Los paraclínicos de ingreso mostraron Leucocitosis (17,820/mm3) a expensas de neutrofilia (79%), bandemia importante (11%), sin anemia (Hb 13.8 g/dL), plaquetas normales (281.000/mm3), los reactantes de fase aguda estaban elevados (proteína C reactiva de 94 mg/L). La función renal fue normal, no había proteinuria, sin embargo el paciente presentó una marcada hipoproteinemia a expensas de las globulinas (proteínas totales 4.8 g/dL, albúmina 3.6 g/dL, globulinas 1.2 g/dL). La cuantificación de inmunoglobulinas séricas totales realizada por nefelometría mostró una hipogammaglobulinemia en rango severo (Inmunoglobulina G total <140 mg/dL, Inmunoglobulina A total 2.9 mg/dL, Inmunogobulina M total <5 mg/dL), con una electroforesis de proteínas compatible con agamaglobulinemia. El mismo día del ingreso se inició inmunoglobulina humana endovenosa (IGIV) a razón de 800 mg/kg de peso. Requirió ventilación mecánica por 5 días, no se documentó ningún patógeno en los cultivos (el paciente recibió antibióticos orales ambulatoriamente) por lo cual completo 14 días de la antibioticoterapia empírica.
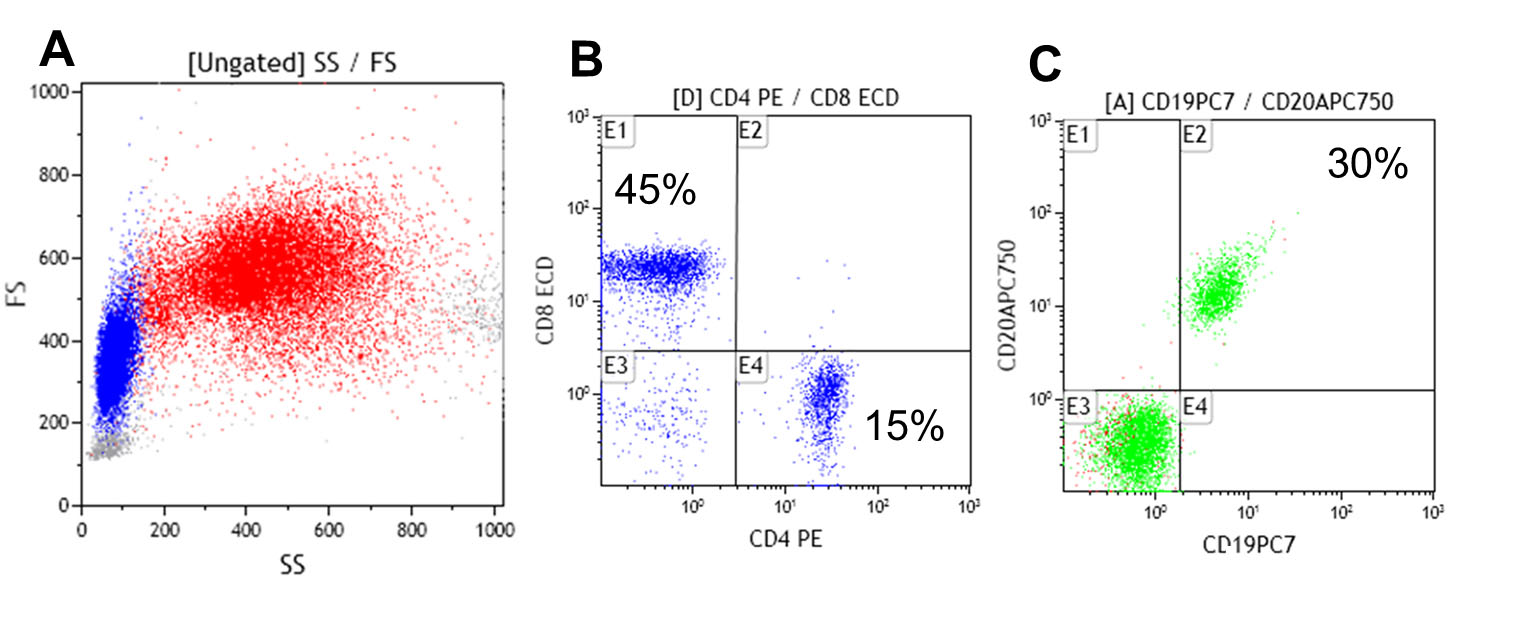
Fue valorado por el servicio de Inmunología previo al inicio de IGIV, encontrando que tanto la respuesta a antígenos de vacunas como a patógenos infecciosos estaba ausente, se determinaron anticuerpos anti-Core y anticuerpos contra el antígeno de superficie del virus de la Hepatitis B (Anti core-VHB, Anti-AgS VHB), ELISA de 4ta generación para VIH, anticuerpos totales contra el virus de la Hepatitis C (VHC), IgM e IgG contra citomegalovirus (CMV), IgG para virus herpes simple 1-2 (VHS), anticuerpos IgG contra tétanos y rubeóla) estos hallazgos demuestran no sólo la deficiencia cuantitativa si no también funcional (específica) de la respuesta inmune humoral. La evaluación de las subpoblaciones linfocitarias en sangre periférica evidenció un conteo absoluto de linfocitos B de 420 células/µL normal (100-500) (Fig. 2C), acompañado de la inversión de la relación linfocitos T CD4+/CD8+ a expensas de la disminución de los linfocitos T CD4+ a 190 células/ µL, (300-1400) (Fig. 2B).
Figura 2.
Cuantificación de las poblaciones de Linfocitos T (CD4+/CD8+ y Linfocitos B (CD19+/CD20+) por citometría de flujo en sangre periférica. Análisis de dot plot por tamaño y complejidad (Forward y Side scattered) del total de leucocitos en A, cuantificación de los Linfocitos T CD4+ y Linfocitos T CD8+ en B y cuantificación de los Linfocitos B totales en C. Nótese la inversión de la relación CD4/CD8, 1:3 (en lugar de 2:1) y el aumento porcentual de los Linfocitos B 30% (6-19%).
En la actualidad el paciente recibe IGIV al 10% a una dosis de 800 mg/Kg cada 4 semanas, recibe profilaxis con TMP/SX 160/800 mg interdiario, completó un ciclo de 22 sesiones de rehabilitación pulmonar y es manejado en conjunto por Inmunología, Neumología y Medicina Interna. La evolución ha sido satisfactoria, durante el último año posterior al inicio de terapia de sustitución con IGIV, el paciente ha presentado sólo un episodio de neumonía que recibió un ciclo corto de Levofloxacina con excelente respuesta, su capacidad pulmonar sin embargo está muy limitada y es oxigeno requiriente. El cuadro clínico del paciente, los hallazgos serológicos, los bajos títulos de inmunoglobulinas séricas y la presencia de linfocitos B (CD19+/CD20+) en sangre periférica configuran el diagnóstico de Inmunodeficiencia Común Variable (IDCV).
Discusión
Las inmunodeficiencias primarias (IDP) son patologías consideradas raras (prevalencias 1:25,000 a 1:50,000), lo cual se traduce en un retraso significativo en el diagnóstico que conlleva al empobrecimiento del pronóstico, aumento de las comorbilidades y empeoramiento clínico, y al aumento de costos al sistema de salud por no ser identificadas a tiempo 4. Tradicionalmente se acepta que las IDP son patologías de la niñez por lo cual la medicina interna pocas veces se ocupa de estas, lo cual contribuye a que los médicos de adultos no estén familiarizados con ésta condición 5.
La Inmunodeficiencia Común Variable (IDCV) es la inmunodeficiencia primaria de relevancia clínica más frecuente en la edad adulta 6,7. Tradicionalmente se acepta que esta patología tiene dos picos de incidencia, uno en la niñez y el otro entre las segunda y tercera décadas de la vida como es el caso de nuestro paciente. El retraso en el diagnóstico es frecuente, ocurriendo a los 2, 15 o inclusive hasta 20 años después de la aparición de los síntomas 7.
La IDCV es una enfermedad muy heterogénea en la cual se han identificado una gran cantidad de mutaciones en genes relacionados con la maduración o activación de los linfocitos B, lo cual finalmente se traducen en una incapacidad tanto cuantitativa como cualitativa para la producción de inmunoglobulinas. En la actualidad se reconocen fundamentalmente dos espectros clínicos: IDCV sin complicaciones que hace referencia al paciente "clásico" con infecciones a repetición sinopulmonares o gastrointestinales y IDCV con complicaciones en el cual se presentan infecciones recurrentes, hay presencia de visceromegalias (hepato/esplenomegalia), adenopatías, autoinmunidad (usualmente citopenias) y malignidad tipo linfomas 3. La presencia de bronquiectasias generalizadas como es el caso de nuestro paciente, obliga a buscar de forma activa y descartar la presencia de inmunodeficiencias primarias, pues se ha demostrado que hasta el 10% de los pacientes con Bronquiectasias (no fibrosis quística) corresponden a deficiencias de anticuerpos especialmente IDCV 8,9.
Los criterios diagnósticos de IDCV fueron establecidos por las sociedades Europea y Panamericana de inmunodeficiencias (ESID/PAGID) en 1999 y siguen vigentes hasta el momento, incluyen: Disminución marcada (al menos 2 desviaciones estándar por debajo del promedio para la edad) de Ig G, A y/o M en suero; ser mayor de 4 años, ser negativo para isohemaglutininas y/o tener pobre respuesta a vacunas, además que se hayan excluido otras causas de hipogammaglobulinemia. El cumplimiento de todos los anteriores criterios, es fundamental para el diagnóstico, previo al inicio de la terapia de sustitución con IGIV debido a que esta terapia modifica los parámetros serológicos hasta seis meses después de su última aplicación 10.
La presencia de linfopenia a expensas de linfocitos T CD4+ puede ser un factor confusor importante, puesto que la mayoría de los médicos tratantes asocian este hallazgo con la infección por VIH/SIDA, como ocurrió en este caso. Es importante resaltar que hasta el 25% de los pacientes con IDCV cursan con conteos de LT CD4+ bajos, sin tener un compromiso de la inmunidad celular.
El objetivo del tratamiento es la sustitución de la respuesta humoral con la administración de Inmunoglobulina humana, con el propósito de disminuir las complicaciones infecciosas, autoinmunes y la aparición de granulomas o malignidad 11. En Colombia están disponibles y autorizados por el INVIMA diferentes presentaciones de inmunoglobulina humana para uso en pacientes con deficiencias de anticuerpos; la (IGIV) que se debe administrar a una dosis entre 400-800 mg/kg cada 3 a 4 semanas dependiendo de los niveles valle y respuesta clínica del paciente, y además contamos con inmunoglobulina humana subcutánea (SubQ) para aplicación semanal entre 100-200 mg/Kg dosis. El uso de profilaxis antibiótica no está claramente establecido, sin embargo se recomienda el uso de macrolidos, quinolonas y para los casos que cursan con conteos de linfocitos T CD4+ bajos se recomienda la profilaxis con Trimetoprim/sulfametoxazol 12,13.
Las IDP en adultos deber ser una posibilidad diagnóstica y en Colombia existen las tecnologías y metodologías para confirmar su diagnóstico 14. El presente reporte de caso pretende llamar la atención sobre una patología que requiere un alto índice de sospecha y la baja prevalencia reportada posiblemente obedece a un subregistro y subdiagnóstico lo cual tiene un impacto en el pronóstico de la enfermedad.