El Síndrome de X Frágil (SXF), es una enfermedad genética heredada a través del cromosoma X, descrito por primera vez en 1943 por Martin y Bell 1. Es la causa principal de déficit cognitivo hereditario y la segunda más prevalente después del síndrome de Down. El SXF, contrario al síndrome de Down en donde la mayoría de los casos son de novo, siempre es heredado y afecta a varios integrantes de la familia ya sea como portadores o con la presentación del SXF. Los hombres afectados tienen un fenotipo clásico de cara alargada, orejas grandes-aladas y macroorquidismo 2. El SXF es causado por una expansión anormal en el número de tripletas CGG ubicadas en la región 5´ UTR corriente abajo del promotor del gen FMR1 (Fragile X Mental Retardation 1 gene), con locus Xq27.3. Esta es una mutación dinámica que expande el número de repeticiones de tripletas CGG con cada generación, pasando del rango de premutación entre 50-200 repeticiones a una mutación completa cuando es heredada de una madre portadora de la premutación a su descendencia 3.
Los afectados con SXF tienen más de 200 tripletas CGG y los portadores de la premutación entre 55 y 200; estos últimos no tienen el fenotipo clásico del SXF pero presentan otros problemas médicos, psiquiátricos y neurológicos. En los últimos 15 años se han hecho múltiples avances en la descripción de las características génicas, función de la proteína codificada por el FMR1, la FMRP; manejo farmacológico y la descripción, en portadores de la premutación, del síndrome de tremor y ataxia asociado al frágil X (FXTAS) y falla ovárica prematura asociada al SXF (FXPOI) 3-5.
El objetivo de esta revisión es aportar a la divulgación del conocimiento sobre el Síndrome de X Frágil, contribuir a su comprensión por parte del personal de salud y así mejorar el diagnóstico y manejo de los pacientes con este síndrome.
1. Epidemiología
La prevalencia actual del SXF determinada a través de pruebas moleculares, se estima en uno de cada 5,000 hombres 6 y a uno de cada 4,000 a 6,000 mujeres a nivel mundial 6,7. Ricaurte es un corregimiento del municipio de Bolívar, al norte del Valle del Cauca, que ha sido identificado como una población con una prevalencia elevada de discapacidad, 39 individuos con retardo mental en 1,124 habitantes 8. A finales de los años 90 se realizó un estudio que encontró que la causa es una enfermedad genética heredable a través del cromosoma X, denominada Síndrome de SXF. En ese estudio se lograron diagnosticar 19 pacientes con el síndrome a través de citogenética, cariotipo con bandas G con déficit de folatos; además se hizo el diagnóstico clínico en 16 más a quienes no se les logró realizar la prueba. Los casos fueron encontrados en 3 familias y se postuló un probable ancestro común dado las características de la fundación del pueblo y patrones migratorios 9. Para el año 1999 se determinó una prevalencia de X Frágil, en Ricaurte, de 1 en 38 hombres y 1 en 100 mujeres, lo que excedió 100 veces lo reportado en la literatura para el síndrome, no encontrándose ninguna población con esa prevalencia.
Por otra parte, existen síndromes y condiciones médicas asociadas al SXF que se manifiestan en portadores de la premutación entre los cuales se encuentran la depresión, ansiedad, cefalea migrañosa, hipertensión, apnea del sueño, enfermedades mediadas por el sistema inmune incluyendo hipotiroidismo y fibromialgia, FXTAS y FXPOI que se han descrito en los últimos 10 años 10. La prevalencia de la premutación en la población general es de 1 en 130-200 mujeres y 1 en 250 hombres 11,12. La ataxia asociado al frágil X ocurre en aproximadamente el 40% de los hombres con la premutación y el 16 % de las mujeres, mientras que FXPOI ocurre en 16-20% de las mujeres con la premutación 2,10.
2. Genómica
El SXF es causado por una alteración en el gen FMR1, con locus Xq27.3. Este gen alberga una repetición de CGG dentro de la región no traducida 5´ (UTR5´). Dependiendo del número de repeticiones se definen cuatro tipos de alelos, los cuales van a tener diferentes manifestaciones clínicas 3,13: los alelos normales, hasta 44 repeticiones de CGG, los alelos con pre-mutación (PM) entre 55 y 200, y los completamente mutados (CM) con más de 200 repeticiones de tripletas. Los alelos denominados alelos intermedios o zona gris, son aquellos que contienen un número de repeticiones entre 45 y 54 y se han propuesto como precursores de alelos PM.
El silenciamiento del gen FMR1 es el resultado de una serie de complejas modificaciones epigenéticas concomitantes a la expansión de la tripleta CGG en el gen 14. Los alelos CM sufren un proceso de metilación las islas CpG dentro del promotor del gen FMR1 y en las repeticiones de CGG 15,16. En pacientes hombres con el alelo CM, toda citosina de las islas CpG se encuentra metilada, contrario a los individuos sanos en quienes hay una ausencia completa de metilación 17. También se ha descubierto dentro del gen FMR1 una secuencia limítrofe metilada que se encuentra entre 650 a 800 nucleótidos corriente arriba de la secuencia de repetición CGG. Esta secuencia limítrofe, por su conformación estructural en la cromatina, delimita las zonas hipermetiladas del genoma, protegiendo al promotor del gen FMR1 de una posible metilación 15,16,18. En individuos con SXF esta secuencia limítrofe se pierde permitiendo la expansión de la metilación hasta el promotor del gen FMR1. Estos hallazgos sugieren que cambios en la secuencia de nucleótidos y su conformación estructural en la cromatina de la secuencia limítrofe favorecerían los cambios epigenéticos que concluirían silenciando al gen FMR1 y por ende previniendo la producción de la proteína FMRP 19.
El SXF es causado usualmente por la metilación y el silenciamiento del gen, asociados a la mutación completa, sin embargo deleciones de la región codificante del gen también pueden conducir a una ausencia de FMRP. Adicionalmente, mutaciones puntuales o desplazamiento del marco de lectura pueden conducir a un déficit funcional de la proteína FMRP y su consecuente fenotipo; sin embargo estos cambios genómicos representan únicamente el 1% de los casos de SXF descritos 20.
La causa del compromiso clínico de los pacientes con la PM es debida a un mecanismo diferente al del SXF; en este caso los niveles de mRNA del gen FMR1 son tan altos que producen toxicidad por RNA. En los portadores de la PM se detectan niveles de mRNA de 2 a 8 veces más altos de lo normal. Este exceso de mRNA puede secuestrar proteínas importantes para la función neuronal en las horquillas que ocurren en las repeticiones de CGG. La toxicidad por RNA induce a la muerte neuronal temprana en cultivo, por ende los portadores de la premutación están en riesgo de desarrollar desordenes del envejecimiento como FXTAS y FXPOI 10.
La FMRP es una proteína de unión a RNA que presenta una preferencia hacia homopolímeros de RNA 21 así como una unión selectiva a subgrupos de transcritos cerebrales 22. La FMRP está involucrada en la regulación de la estabilidad del RNA, el transporte subcelular y la traducción de mRNAs neurales que codifican proteínas involucradas en el desarrollo de la sinapsis, la plasticidad neural y el desarrollo cerebral 23-25. Varios estudios han revelado que en la ausencia de esta proteína, una amplia gama de mRNAs neurales están alterados, aumentando la síntesis proteica neural y dando como resultado la producción de una dismorfogénesis espinal acompañada por un claro desbalance excitación/inhibición (Glutamato/GABA), fenómenos presentados en los pacientes con SXF 26,27. Esta dismorfogénesis espinal, juega un papel importante en el cuadro clínico de los pacientes con SXF debido a la debilidad de las conexiones sinápticas que conducen a déficits intelectuales y alteración del comportamiento. La ausencia de FMRP produce la disminución de múltiples sistemas de neurotransmisores y se aumenta la producción de proteína en el hipocampo y en todo el cerebro en general 3.
3. Heredabilidad
La herencia del SXF no cumple con los patrones clásicos mendelianos. La heredabilidad depende del número de repeticiones de la tripleta CGG en el promotor del gen FMR1 28. El paso de alelos PM a CM ocurre por fenómenos expansivos durante la transmisión materna del cromosoma X, que presenta la PM, a los hijos 2. A su vez este fenómeno expansivo no ocurre en la transmisión del cromosoma X con PM de hombres a sus hijas 28. De manera que todas las hijas de hombres con la PM serían portadoras obligadas de la PM, sin embargo estas mujeres tendrían un riesgo de 50% de tener hijos con SXF.
3.1 Dinámica de la mutación
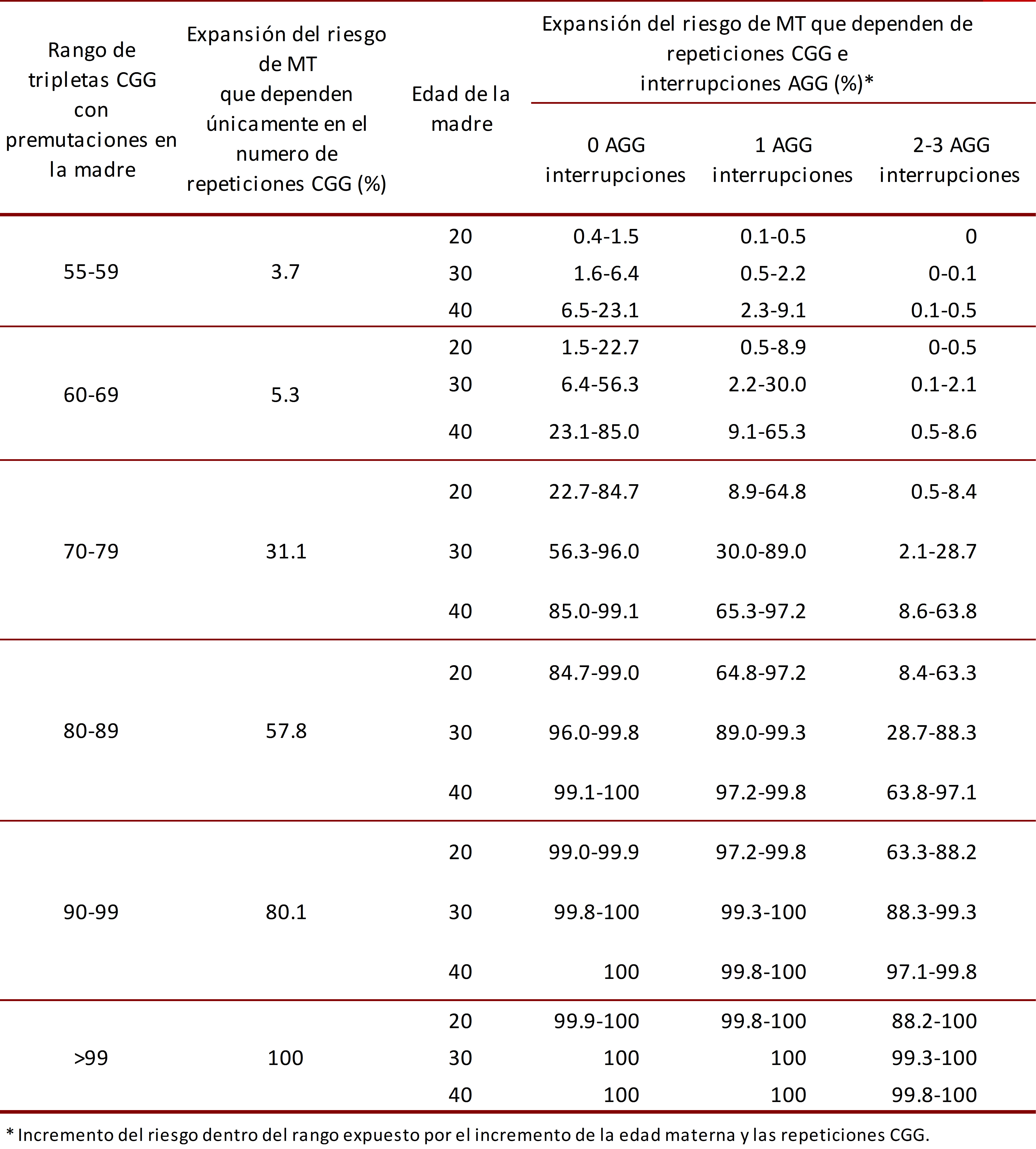
El riesgo de la transición de alelos PM a alelos CM, en la descendencia, depende del número de repeticiones de la tripleta CGG en el alelo PM, siendo el riesgo de transmisión cerca del 100% para alelos PM que presenten más de 99 repeticiones 29, 30. La expansión de PM a CM en meiosis puede ocurrir en alelos con al menos 56 repeticiones de CGG 31; la probabilidad de que esto ocurra depende del rango de repetición de tripletas en el que se encuentre el paciente con PM (Tabla 1) repeticiones 29, 30. Normalmente existe una tripleta AGG que interrumpe las repeticiones de CGG entre cada 9 a 10 tripletas en el gen FMR1. Estas tripletas AGG actúan como un anclas, las cuales pueden modificar el riesgo de expansión del número de repeticiones CGG al ser heredadas de la madre con la PM a sus hijos. Las mujeres con la PM y dos anclas de AGG tienen menor riesgo de expansión a CM en comparación con las mujeres con la PM y sin ninguna interrupción de AGG) repeticiones 29, 30.
Los alelos denominados alelos intermedios o zona gris, son aquellos que poseen un número de repeticiones entre 45 y 54, siendo propuestos como precursores de alelos PM. El paso de alelos intermedios a PM ocurre por fenómenos de inestabilidad meiótica tanto paterna como materna 32.
3.2 Riesgo de herencia y de recurrencia
Hombres. La mayoría de los hombres con CM usualmente no se reproducen, solo el 1% han reportado tener hijos 2. Los pacientes con CM y SXF tendrán un 100% de probabilidad de heredarle a sus hijas la PM lo que las hace portadoras que usualmente no presentan retraso mental. Hay una pérdida de la CM en la producción de los espermatozoides y solo la PM se puede heredar. Por otro lado, todos los hijos varones recibirán el cromosoma Y del padre con la CM, de manera que no heredaran la mutación del gen FMR1 presente en el cromosoma X. De otro lado, los hombres con la PM heredaran únicamente la PM a sus hijas, siendo el numero de repeticiones CGG relativamente estable 2,31.
Mujeres. Las portadoras de la PM pueden expandir su alelo a CM dependiendo del número de tripletas CGG, número de interrupciones AGG y edad materna (Tabla 1). El impacto de las interrupciones AGG es atribuido a la reducción del riesgo de deslizamiento de la ADN polimerasa en la replicación en meiosis. De este modo, las interrupciones dan estabilidad a la transmisión del gen pero no afectan la traducción y transcripción del mismo) 29, 30.
4. Características clínicas del Sindróme Fragil X
El fenotipo cognitivo y emocional de los pacientes con SXF va a depender de la cantidad de proteína FMRP que se esté produciendo, acorde con el número de tripletas y el grado de metilación que presente el gen FMR1 en cada uno de los pacientes. Dependiendo de la concentración de proteína FMRP se desarrolla un espectro fenotípico. Cuando los niveles de FMRP no son tan bajos se presenta una leve afectación emocional y dificultades en el aprendizaje, acompañado de un coeficiente intelectual normal. Si la producción de esta proteína disminuye o desaparece por completo se desarrolla un déficit cognitivo severo produciendo retardo mental 2.
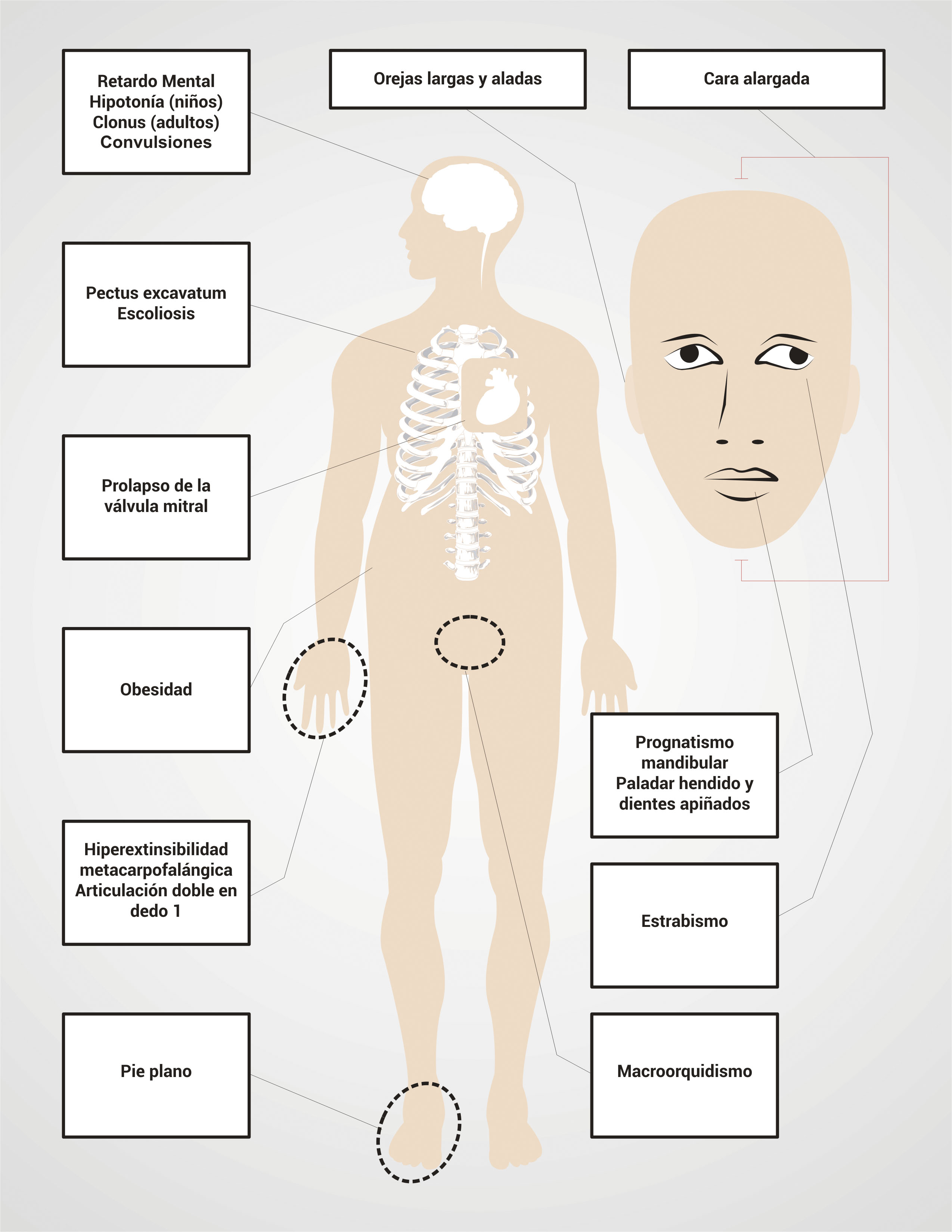
Las características físicas particulares de los pacientes con SXF también dependen de la producción de FMRP. Así, el 80% de los pacientes con SXF tendrán una o más de las características faciales comunes (Tabla 2 y Fig. 1) 2. El diagnóstico de SXF se sospecha en hombres con retardo mental, características faciales particulares como cara larga, mandíbula y orejas grandes, estas últimas haladas y macroorquidismo; esta característica permite distinguir a los pacientes con SXF después de la adolescencia de los pacientes con retardo metal no asociado al SXF 2,20. Sin embargo, debido a que algunas veces las características físicas no son obvias o no están presentes, especialmente en el caso de niños pequeños, todos los individuos con discapacidad intelectual o autismo deben ser sometidos a un examen de DNA para frágil X.
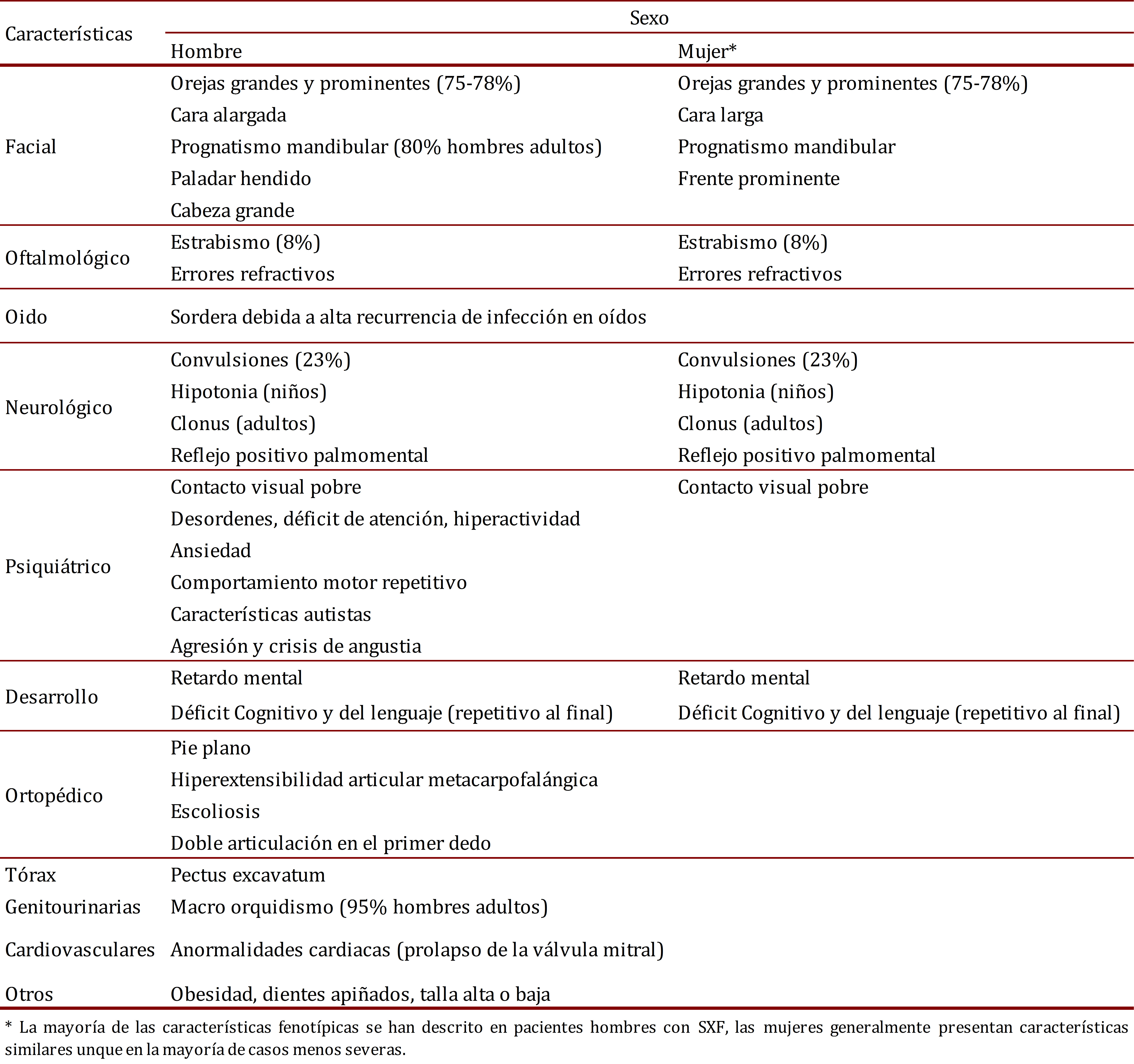
Las mujeres con la CM y SXF van a presentar un rango de compromiso fenotípico mucho más amplio que el de los hombres, dependiendo de la tasa de activación (TA) en sangre periférica del cromosoma X mutado (TA= porcentaje o rango de células con el alelo normal presente en el cromosoma X activo; así, un alto TA corresponde a mayores niveles de FMRP producidos por el alelo normal de FMR1). La severidad del fenotipo en las mujeres puede variar desde la presencia de características físicas (Tabla 2 y Fig. 1) y retraso mental hasta la ausencia del fenotipo físico con leves problemas en el aprendizaje. 70% de las mujeres con la CM presentan algún grado de déficit cognitivo 2.
Por el contrario los portadores de la PM usualmente presentan un Coeficiente Intelectual normal con leves o sin características físicas; sin embargo, el 20% de las mujeres portadoras de la PM presenta FXPOI o FXTAS, y el 40% de los hombres portadores desarrolla FXTAS. El FXTAS es un desorden neurodegenerativo progresivo10, de inicio tardío, que ocurre entre los portadores de la premutación, caracterizada por déficits neurológicos que incluyen temblor progresivo de intención, ataxia cerebelar, parkinsonismo, neuropatías y disfunción autonómica 33-35. Por su parte, FXPOI (indicada por la interrupción de la menstruación antes de los 40 años de edad) afecta únicamente a las mujeres produciendo una serie de complicaciones en la reproducción y fertilidad, ya que este síndrome se caracteriza por irregularidades en el ciclo menstrual, infertilidad y deficiencia hormonal ovárica 36.
5. Diagnóstico
El diagnóstico de SXF inicialmente se realizaba a través del cariotipo al observarse en la microscopia de luz un estrechamiento del extremo distal del brazo largo del cromosoma X en la banda 27.3 (Xq27.3 23.8). Estos hallazgos se encuentran en diferentes cromosomas y se conocen como puntos frágiles, de allí el nombre de SXF 2.
Actualmente se dispone de pruebas moleculares para el diagnóstico del SXF que son más sensibles y específicas que el cariotipo 11. Además de diagnosticar a los pacientes que padecen el síndrome y tienen la CM, estos exámenes también permiten la identificación de portadores de la PM; individuos que generalmente tienen un coeficiente intelectual normal, pero que presentan un alto riesgo de tener hijos con el SXF. También hay pruebas moleculares que permiten cuantificar RNA mensajero (mRNA) y la cantidad de la proteína FMRP, las cuales se realizan para un mejor entendimiento de la fisiopatología de la enfermedad al correlacionarlas con el fenotipo de los pacientes CM y portadores de la PM 2. Las pruebas de rutina para el diagnóstico de SXF son la Reacción en Cadena de la Polimerasa (PCR) y el Southern blot que permiten determinar el número de tripletas CGG y el estado de metilación del gen FMR1.
La PCR a través de la utilización de cebadores específicos para el gen FMR1, permite la amplificación de la zona que contiene la repetición de CGG y puede identificar los pacientes con un alelo expandido de FMR1, particularmente en el rango de la CM pero también en el de la PM 37. El análisis por Southern Blot es utilizado para caracterizar mejor alelos en el rango de la PM y para determinar el estado de metilación en las CM 38. Sin embargo con las nuevas técnicas de PCR que utilizan dobles cebadores para un PCR anidado, se logran cuantificar repeticiones de las tripletas CGG e identificar no solamente la CM sino pacientes con la PM 37,39.
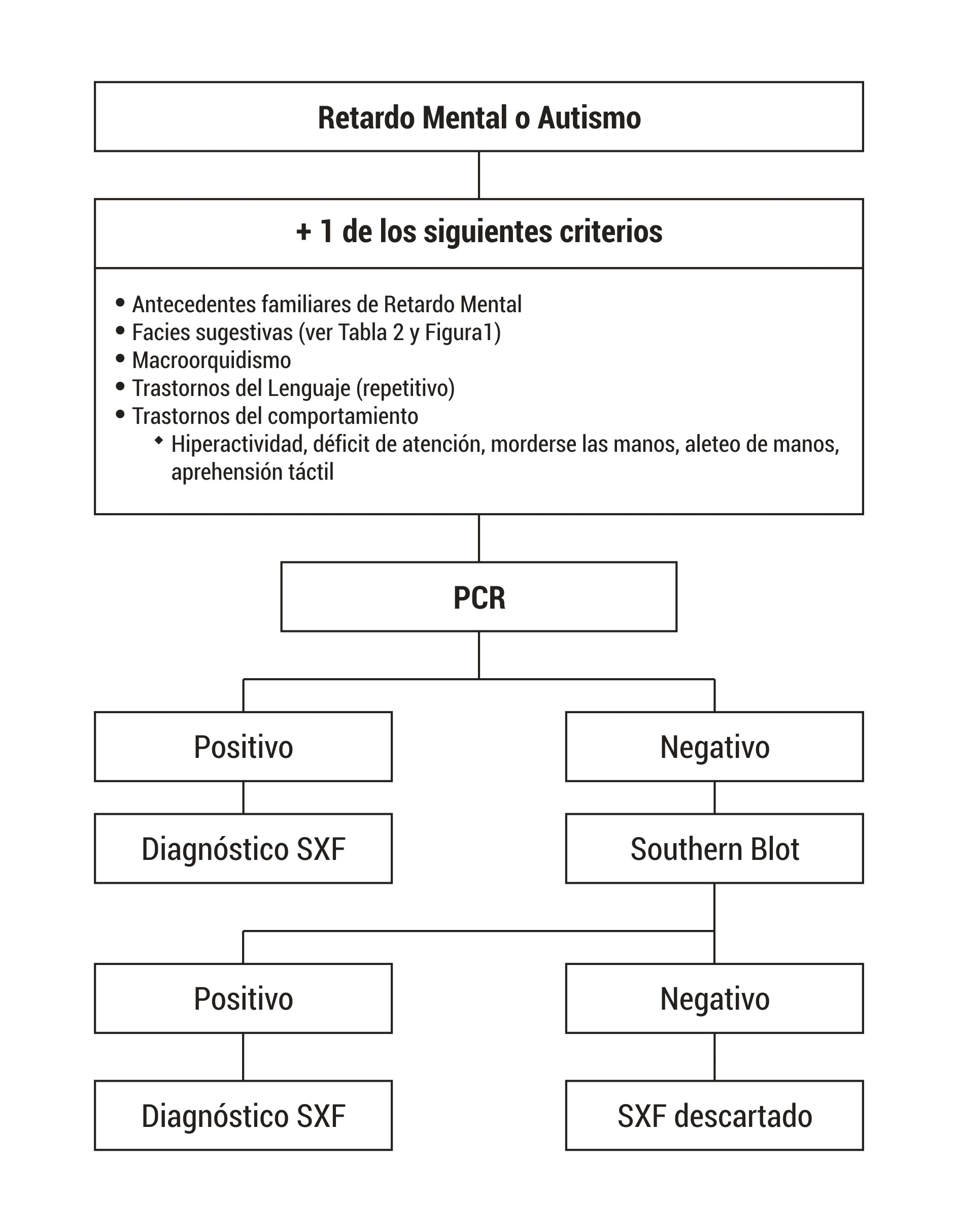
Las pruebas para realizar el diagnóstico de SXF se deben solicitar a pacientes con déficit intelectual y/o autismo y además alguna de las siguientes características: fisionomía especial como orejas grandes y aladas, cara alargada, entre otras; historia familiar de déficit intelectual, autismo, macroorquidismo (Fig. 2).
Una vez se determina que el paciente tiene la mutación completa o la premutación para el SXF se deben realizar pruebas moleculares a los familiares que sean sospechosos de ser portadores a través del heredograma. También serían candidatos a realizarles pruebas moleculares para SXF a los familiares que tengan tremor, ataxia, síntomas neurológicos o falla ovárica prematura 40.
El diagnóstico de SXF se puede hacer incluso en fetos de mujeres portadoras o con SXF en estado de embarazo utilizando las pruebas moleculares mencionadas anteriormente (Southern blot y PCR) con una muestra de vellosidades coriónicas para la realización del análisis 40.
6. Diagnóstico diferencial
El diagnóstico diferencial incluye los síndromes de Sotos, Prader-Willi y Klinefelter; con los cuales se comparten algunas características particulares. Para cada uno de estos síndromes existen pruebas moleculares específicas con las que se puede confirmar el diagnóstico, estas se solicitarán según los hallazgos fenotípicos y el análisis clínico (Tabla 3) 2,41,42. Los hallazgos clínicos más frecuentes en estos síndromes y a contrastar con el SXF son:
Síndrome de Sotos: retraso mental, talla elevada, macrocefalia, epilepsia.
Síndrome de Prader-Willi: retraso mental, obesidad, baja talla, hipogenitalismo. Un subgrupo de pacientes con SXF tendrá el fenotipo de Prader- Willi pero no presentará la deleción en la región 15q11-13 a pesar de que los niveles de la proteína CYFIP de esta región sean bajos.
Síndrome de Klinefelter: Talla alta, hipogenitalismo, deficiencia mental (20%).

Otros síndromes menos frecuentes, con una prevalencia menor a uno en 50,000, presentan mecanismos genéticos, fisiopatogénicos y un fenotipo similar al SXF. Entre estos encontramos los síndromes FRAXE, FRAXF y FRAXD con alteraciones en los genes FMR2, FAM11A y FRAXD respectivamente. Si el fenotipo es altamente sugestivo de SXF y el Southern Blot es negativo, pruebas moleculares para estos genes deben ser consideradas.
7. Tratamiento
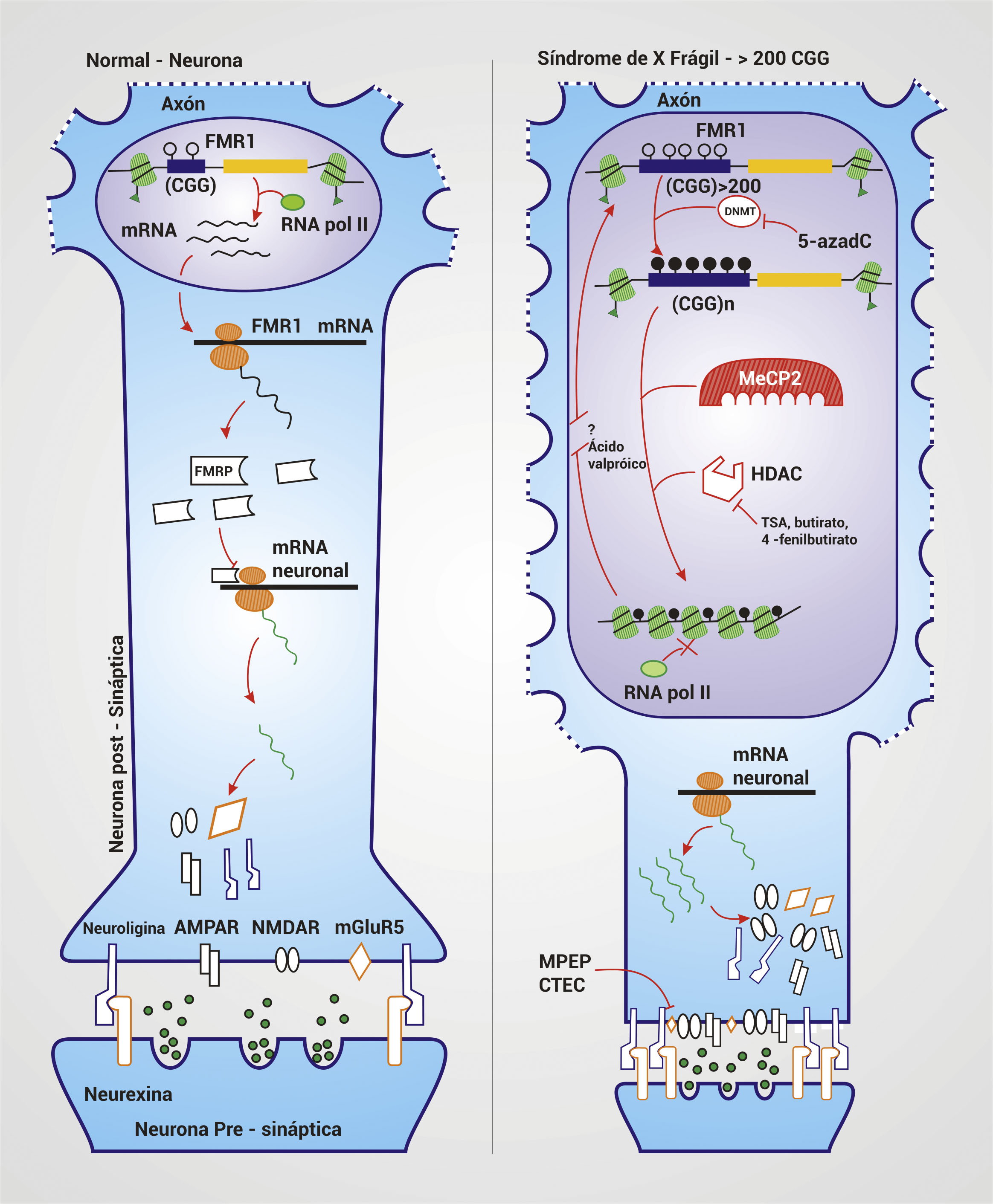
Múltiples estudios se han realizado en búsqueda del desarrollo de tratamientos específicos para el SXF, que puedan mejorar algunos síntomas y la calidad de vida de los afectados. Estas investigaciones han tenido diferentes enfoques según sus objetivos, ya sea la restauración de la función del gen FMR1 o el tratamiento de los síntomas asociados a esta enfermedad. Los mecanismos de acción de los fármacos utilizados, se enfocan en moduladores epigenéticos, sistema glutamatérgico y regulando la traducción de mRNAs blanco de FMRP (Fig. 3).
Figura 3.
Blancos terapéuticos y fisiopatogénesis en SXF. En una neurona con un rango de repeticiones normal de CGG en el gen FMR1, hay una conformación estructural laxa de la cromatina que permite el ingreso de la maquinaria de transcripción de genes, debido a la acetilación de las histonas H3 y H4. Esta conformación permite la producción normal de la FMRP, proteína que inhibe la traducción de mRNAs neuronales que codifican para proteínas como la Neuroligina, receptor AMPA (AMPAR), receptor NMDA (NMDAR) y receptor mGluR5 entre otras. La expansión >200 repeticiones de CGG lleva al reconocimiento de las islas CpG por DNA Metil Transferasas (DNMT) que añaden grupos metilo a las citosinas de la secuencia e incluso del promotor lo que silencia el gen. Además, estas citosinas metiladas son reconocidas por MeCP2, la cual atrae a Deacetilasas de Histonas (HDAC) que retiran los grupos acetil de las histonas H3 y H4 cercanas a la secuencia metilada llevando a una condensación de la cromatina en el segmento afectado. Juntos estos cambios impiden la producción de FMRP, permitiendo la excesiva traducción de mRNAs neuronales lo que causa hiperexcitación neuronal, dismorfogénesis neuronal y las sucesivas manifestaciones neurológicas. El 5-azadC es un fármaco que inhibe la DNMT impidiendo la metilación de las citosinas. Debido a que los fármacos TSA, butirato y 4-fenilbutirato inhiben las HDAC, estos actúan sinérgicamente con el 5-azadC. El ácido valpróico, por su parte, parece reactivar genes silentes sin que se haya dilucidado por completo el mecanismo por el cual lo logra. Los inhibidores del receptor mGluR5 permiten reducir su hiperexcitación lo cual genera la sobreproducción de este receptor y las consecuencias de ésta.
Aunque en cultivos celulares, modelos del SXF, el ácido fólico disminuyó la observación de puntos frágiles en el cromosoma X, este medicamento no tiene ese mismo resultado in vivo. Su efecto benéfico puede deberse al papel que tiene este micronutriente en la metilación e hidroxilación de neurotransmisores, reacciones implicadas en su síntesis y metabolismo 2. Los primeros ensayos clínicos con este suplemento en la década de 1980 reportaron una mejoría similar a la obtenida mediante el uso de estimulantes; específicamente en la coordinación motora, el lenguaje y el discurso. Sin embargo, su efectividad ha sido difícil de demostrar en estudios controlados considerándose con una efectividad limitada y con contraindicación en pacientes con SXF y convulsiones2,43.
El ácido valpróico, que actualmente se conoce como un reactivador de genes silentes 3,44, parece ser un reactivador débil de alelos CM 3,45. Se ha encontrado que su administración en pacientes con SXF mejora la hiperactividad y el déficit de atención. En pacientes con convulsiones tónico-clónicas generalizadas, crisis de ausencia y convulsiones parciales es el medicamento de elección. Dado que aumenta los niveles de GABA y disminuye los de Dopamina, es un excelente estabilizador del afecto y se puede utilizar en casos de trastorno bipolar en adultos e inclusive en niños 2,3,46.
La L-Acetil-carnitina también parece inhibir la formación de los puntos frágiles en el cromosoma X. Su administración a pacientes con SXF llevó a una mejora significativa del trastorno por déficit de atención e hiperactividad (TDAH), aunque el estado de metilación del gen FMR1 no cambió y su expresión tampoco mejoró 2,3. El uso de este medicamento pudiera ser indicado aunque los estudios no son concluyentes.
Los mejores medicamentos disponibles para el tratamiento de los síntomas de TDAH en pacientes con SXF son los estimulantes como por ejemplo preparaciones de metilfenidato o sales mezcladas de anfetaminas. Sin embargo, estos fármacos no deben ser administrados a niños menores de 5 años ya que pueden causar irritabilidad 47. En el caso de que se presenten síntomas severos de TDAH en niños menores a 5 años, estos se deben tratar con guanfacina o clonidina. La clonidina también puede ser empleada en el tratamiento de trastorno del sueño, el cual es muy común entre los niños jóvenes con SXF. Como alternativa para mejorar el sueño a estos niños se puede utilizar melatonina en una dosis de 1 a 3mg a la hora de dormir 48.
El uso de antagonistas mGluR, como el 2-metil-6-(feniletinil) piridina (MPEP) y el CTEC, ha dado como resultado un rescate de la morfología de las espinas dendríticas, síntesis proteica, la atrofia en el hipocampo, y parcialmente el macroorquidismo en modelos animales 3,49. Sin embargo, el uso de antagonistas mGluR5 no ha demostrado eficacia en adolescentes y adultos con SXF en diferentes estudios controlados.
Uno de los grandes intereses y blancos farmacológicos para el tratamiento de SXF es restaurar la actividad del gen FMR1 disminuyendo la metilación del ADN y alterando la acetilación de las histonas H3 y H4. El uso del fármaco 5-azadC (inhibidor de metiltransferasas), en conjunto con inhibidores de deacetilasas de histonas (como TSA, butirato y 4-fenilbutirato) han probado actuar sinérgicamente para este fin. Ambos fármacos se han utilizado únicamente in vitro, esto debido a que in vivo existe riesgo potencial de inducir apoptosis celular 3.
Por otro lado, al parecer la FMRP regula la estimulación sináptica 50, por lo tanto, al estar ausente la FMRP en el SXF habría una sobreproducción de la metaloproteinasa 9 (MMP9) en respuesta a estimulaciones sinápticas normales 51, lo queha sido asociado con varias condiciones patológicas, particularmente convulsiones y accidentes cerebrovasculares 52. La forma activa de MMP9 regula el ambiente pericelular a través del clivaje de componentes proteicos. Esta función de MMP9 ha sido asociada con cambios sinápticos que ocurren en los procesos de aprendizaje y memoria 53. Se ha demostrado que el tratamiento con minociclina por 3 meses en algunos casos de pacientes con SXF disminuye la actividad de la MMP9 sin influencia de la edad, ni de la dosis administrada de este medicamento 54. Un ensayo clínico controlado demostró la eficacia de la minociclina (dosis de 25 a 100 mg por día) en el mejoramiento del comportamiento, el ánimo y la ansiedad de los niños entre 3.5 a 16 años 55.
La minociclina se puede usar clínicamente en el tratamiento de niños y adultos con SXF y aunque su uso en niños menores de 8 años puede producir un oscurecimiento dental de los dientes; el mayor efecto de este fármaco sobre el fortalecimiento de las conexiones sinápticas y el aumento del desarrollo cognitivo se produce cuando se administra en niños jóvenes. El médico tratante debe discutir los efectos secundarios de la minociclina con la familia antes de hacer la prescripción del medicamento, teniendo en cuenta que el oscurecimiento dental puede ser tratado cosméticamente cuando el niño sea mayor. La minociclina puede producir diarrea la cual usualmente mejora con el uso de probióticos. Debido a que en raras ocasiones este medicamento puede causar una elevación de los anticuerpos antinucleares (ANA), la presencia de sarpullido, inflamación de las articulaciones, problemas visuales o cefalea severa secundario a la administración de la minociclina, es motivo suficiente para descontinuar el tratamiento 55.
Debido a la alta frecuencia de trastornos del afecto, ansiedad y desordenes del comportamiento en los pacientes con SXF y portadores de la PM, los inhibidores selectivos de la recaptación de serotonina (ISRS) han sido utilizados para tratar de manera específica y efectiva la agresión, ansiedad, depresión y demás trastornos 2. El uso de la sertralina en SXF es un tema actual de investigación, algunos artículos han reportado su efecto positivo sobre el desarrollo del lenguaje en niños de 2 a 6 años con SXF entre otras virtudes 56. No obstante, en un 20% de los casos los ISRS pueden empeorar la agresividad de los pacientes debido a hiperactivación especialmente si la dosis es alta. De manera que si la agresividad se desarrolla en estos pacientes secundario al uso de ISRS, la dosis debe ser disminuida o descontinuada y reemplazado por un estabilizador del ánimo como aripiprazol o risperidona. El litio también es un excelente estabilizador del animo en SXF y es un tratamiento dirigido para SXF 47.
Además de todas estas posibilidades farmacológicas de tratamiento, diversos autores hacen hincapié en la necesidad de realizar terapias cognitivas y de comportamiento, además de estímulos educacionales, en estos pacientes para así fortalecer sus habilidades sociales, de lectura, comportamientos adaptativos y redes de apoyo 57-61. Los niños con SXF generalmente tienen una gran habilidad con los computadores, de manera que el uso de aplicaciones en estos equipos que se enfoquen en los déficits de las habilidades sociales, de lenguaje, lectura o matemáticas puede ser de gran ayuda para maestros, terapeutas o padres.
8. Conclusión
En Colombia no se ha descrito la prevalencia de SXF, pero existen muchos pacientes con la mutación completa y portadores de la premutación, incluso un foco endémico en Ricaurte, Valle del Cauca. El conocimiento de la genómica, la fisiopatología de la enfermedad, las funciones de la FMRP en el sistema nervioso central, la heredabilidad y hallazgos fenotípicos en los portadores ha aumentado en la última década. Esto ha permitido estimular múltiples investigaciones, especialmente en intervenciones farmacologicas con tratamientos dirigidos que puedan revertir la neurobiología del SXF. No obstante, los medicamentos que aumentan los niveles de la FMRP o la expresión del gen FMR1 no se han usado in vivo por razones de seguridad; otros medicamentos como ácido valpróico, sertralina, minociclina entre otros, han dado buenos resultados en síntomas específicos en los pacientes con SXF aunque deben ser utilizados de acuerdo a la respuesta individual del paciente.