Introducción
El Síndrome de Apert (SA) es un desorden autosómico dominante con prevalencia de 1-9/100,000 nacimientos, es caracterizado por la sinostosis de suturas craneales y acrocefalia, incluyendo braquicefalia, hipoplasia medio-facial, y sindactilia de manos y pies 1.
Durante el desarrollo craneofacial en diferentes etapas de la formación embrionaria, las vías de señalización del factor de crecimiento de Fibroblastos (FGF), sus receptores (FGFR), y específicamente el receptor del factor de crecimiento de fibroblastos tipo 2 (FGFR2), regulan el balance entre la proliferación y la diferenciación de células osteogénicas en la cresta neural. Luego, en el desarrollo, estas vías se encuentran involucradas además en la formación del cartílago, de los huesos de la bóveda craneal y del maxilar, así como en la migración de las placas que permiten la formación del paladar y los labios 2. En 1995, Wilkie et al., demostraron la presencia de mutaciones en el gen FGFR2 en pacientes con SA, siendo P253R y S252W las mutaciones más frecuentes 3.
El gen FGFR2, localizado en el cromosoma 10, locus 10q26, codifica para un receptor transmembranal que posee una región extracelular compuesta de tres dominios similares a las inmunoglobulinas IgI, IgII e IgIII, con un segmento transmembranal hidrofóbico y un dominio citoplasmático de tipo tirosin-quinasa 1. Los dominios Ig corresponden a la expresión de los exones 8, 9 y 10, en donde la mayoría de los pacientes (25-75%) registran mutaciones 4.
Otras mutaciones localizadas en esta región han sido identificadas en los Síndromes de Crouzon y de Pfeiffer, con craneosinostosis, y con la presencia o ausencia de labio y paladar hendido 5.
En este reporte se presentan dos casos con SA con hallazgos interesantes que podrían brindar información para el conocimiento de esta patología. El objetivo para la familia 1 fue identificar a través del análisis molecular, el caso índice y su hermano con diagnóstico de labio y paladar hendido (LPH), tienen la misma mutación, ya que se considera una posible relación entre mutaciones en el FGFR2 y los fenotipos SA y LPH. El objetivo para la familia 2 es identificar la mutación.
Materiales y Métodos
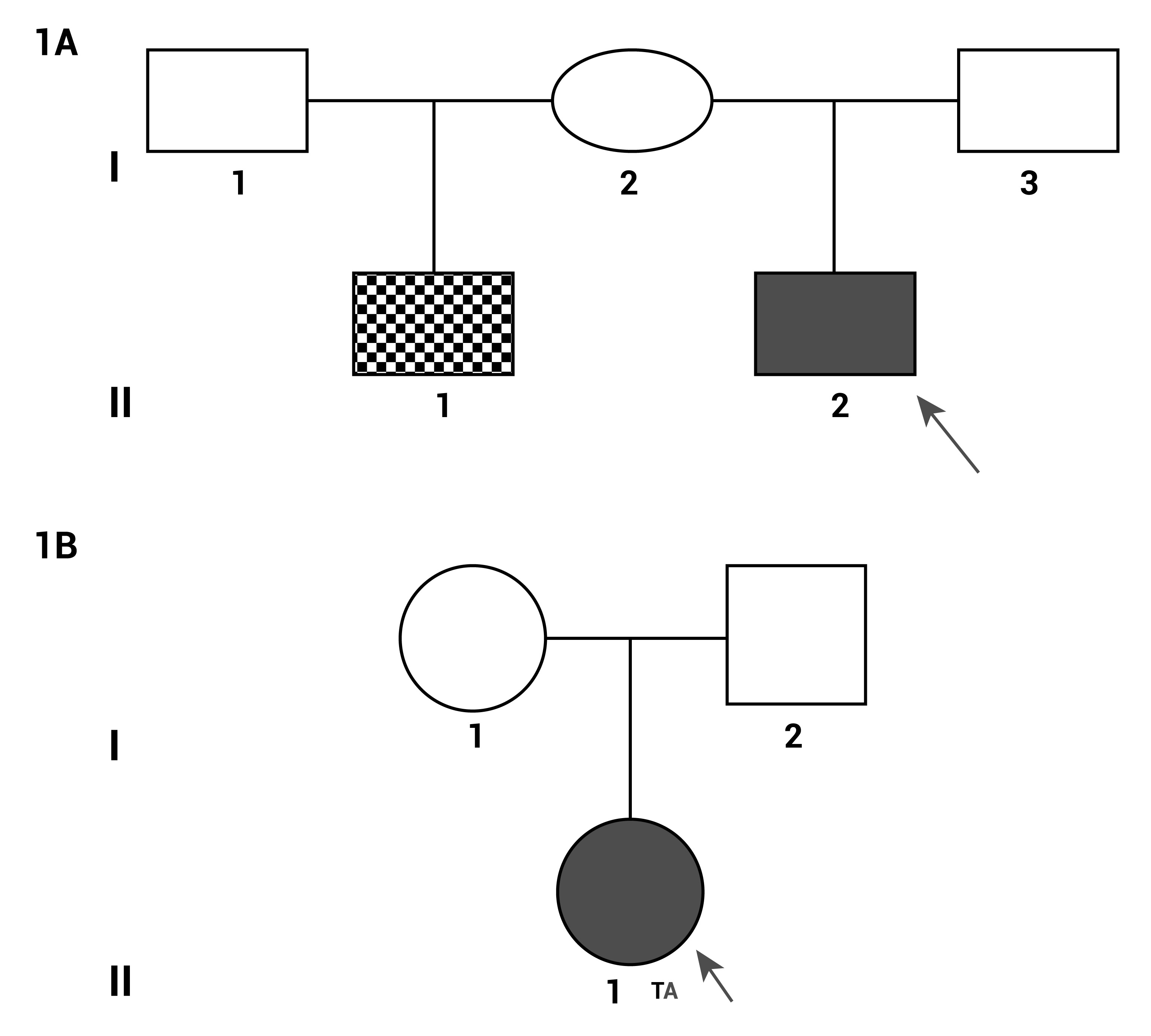
Se analizaron dos familias con un caso índice de SA, familia 1: Compuesta por la madre sana sin características de craneosinostosis, el caso índice de SA y su medio hermano con LPH (I 1, II 2 y II 1) (Fig. 1A). Familia 2: Compuesta por padres sanos y el caso índice de SA (I 1, I 2 y II 1) (Fig. 1B).
Figura 1.
1A. Genealogía de la familia 1. I, 2: Madre sana. II, 1: Medio hermano con LPH. II, 2: Caso índice de SA. 1B. Genealogía de la familia 2. I, 1 y I, 2: Padres sanos. II, 2: Caso índice de SA.
Los paciente de ambas familias fueron diagnosticados con SA y remitidos por las consultas de genética y cirugía plástica al laboratorio de Genética y Biología Molecular, en donde se firmaron los consentimientos informados autorizando el análisis molecular y la publicación de los resultados genéticos.
A partir de las muestras de sangre se realizó la extracción del ADN, por medio del método Salting-out, seguido esto de la amplificación por PCR de los exones 8, 9 y 10 del gen FGFR2. Los primers para la amplificación de los exones fueron diseñados usando la secuencia de referencia obtenida de la NCBI, NW_004078068. La amplificación del exón 8 (E8) se realizó usando el E8-primer forward 5`cccatgaaggagaccccagttg3´ y el primer reverse 5`cattctcccatccccactccctc3´, para E9-forward 5´aatgctaagaccttcctggttgg3´ y reverse 5´cagtctcccaaagcaccaagtc3´ y para E10-forward 5ágccttctcagatggagccagg3´ y reverse 5´gagtctccatcctgggacatgg3´. Las condiciones de amplificación fueron: 95º C por 3 min, seguido de 35 ciclos a 95º C por 1 min, 58º C por 1 min y un paso final a 72º C por un min. Los productos amplificados fueron evaluados por secuenciación y los datos obtenidos analizados usando los programas BioEdit y Blast.
Resultados
Familia 1
Consiste en tres personas, una madre sana (I,1) y dos hijos, el mayor con diagnóstico de LPH (II,1) y el caso índice con SA (II,2) (Fig. 1A).
I, 2: La madre de los pacientes afectados II,1 y II,2 sin características clínicas de craneosinostosis o LPH. El análisis molecular no evidencia cambios en la secuencia de referencia en los exones 8, 9 y 10 (Fig. 2A).
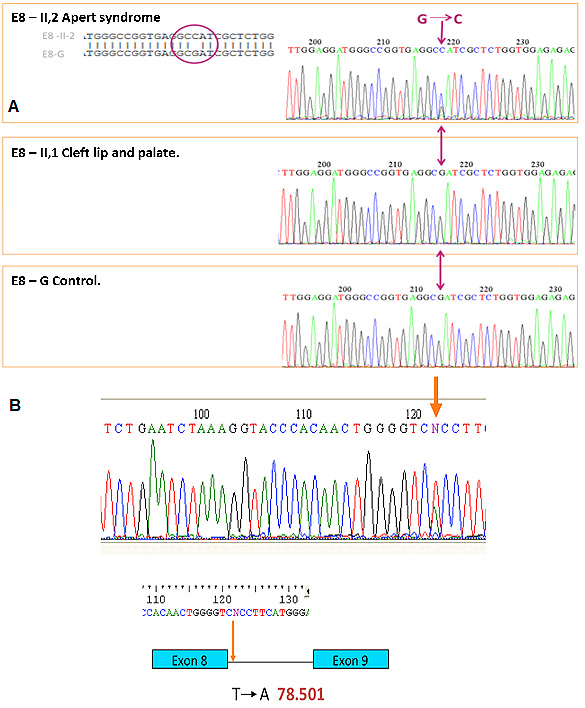
Figura 2.
A. Familia A. I, 2: Madre sana sin mutación, II, 1: Paciente masculino con Labio y paladar hendido (LPH) sin mutación, y II, 2: Paciente masculino con Síndrome Apert quien tiene la mutación S252W en el exón 8. B. Familia B. II, 1: Paciente masculino con Síndrome Apert, quien tiene la variante T78.501A en la secuencia del intrón 8.
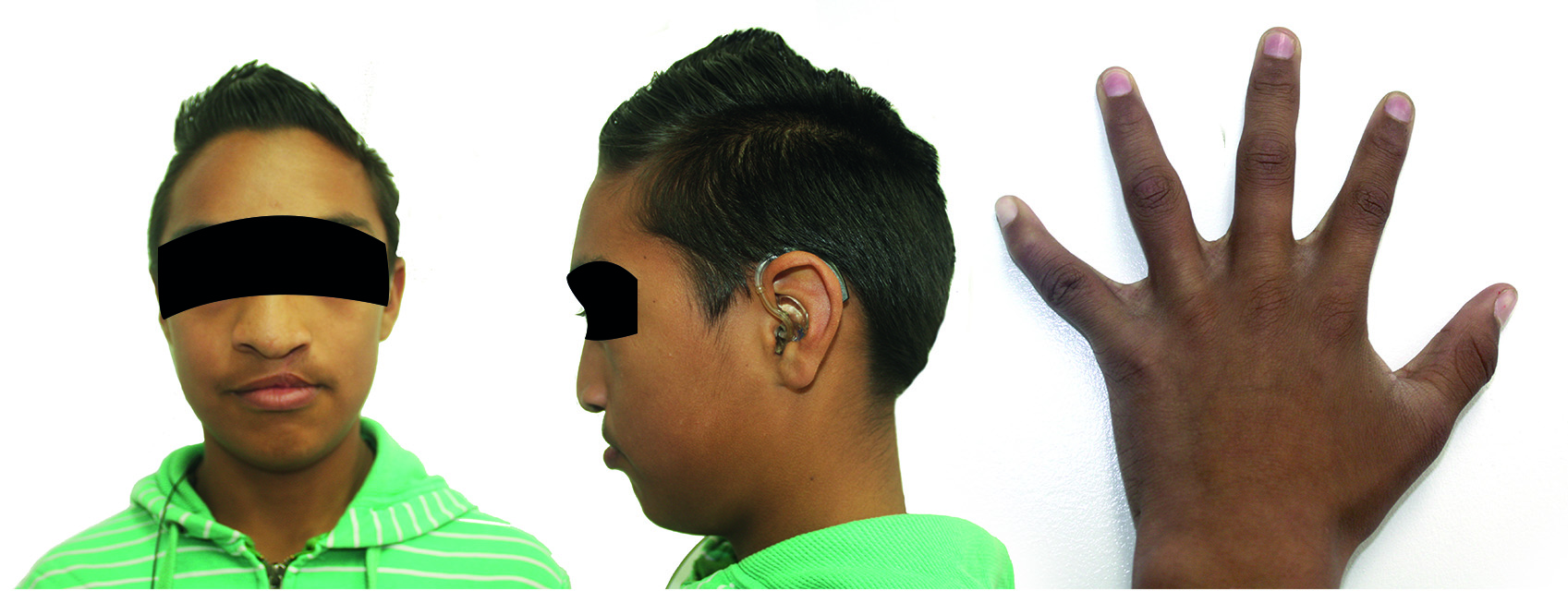
II, 1: Paciente masculino diagnosticado con LPH, acrocefalia, aplasia cutis, hipertelorismo, orejas displásicas y ensanchamiento metafisiario (Fig. 3). Al examen clínico se consideró una posible craneosinostosis. Sin embargo, luego del examen molecular, no se lograron identificar cambios en la secuencia de los exones 8, 9 y 10 (Fig. 2A).
Figura 3.
Paciente masculino (II, 1) diagnosticado con LPH, acrocefalia, aplasia cutis, hipertelorismo, orejas displásicas y ensanchamiento metafisiario.
II, 2: Paciente masculino producto de la segunda gestación. La madre del paciente presentó amenaza de parto pretérmino por lo cual recibió tratamiento para la maduración pulmonar fetal, y tuvo parto vaginal eutócico a las 37 semanas de gestación. Al nacimiento se obtuvo un producto de 3,750 gr de peso y 50 cm de longitud. Al mes de edad el paciente fue diagnosticado con SA y características craneofaciales tales como hipoplasia medio-facial, implantación baja de orejas, puente nasal deprimido, turricefalia, y sindactilia ósea en manos y pies (Fig. 4A), sin historia familiar o consanguinidad.
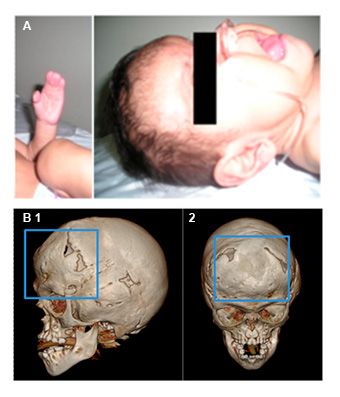
Figura 4.
A. Paciente masculino (II, 2) diagnosticado con Síndrome Apert. Características craneofaciales incluida la hipoplasia medio-facial, implantación baja de orejas, depresión del puente nasal, turricefalia y sindactilia ósea se manos y pies. B. Tomografía computarizada de cráneo (TC) con modelo de reconstrucción tridimensional del paciente (3D) II, 2, Familia 1. Sinostosis de las suturas (recuadro azul) e hipoplasia medio-facial evidente en las imágenes lateral (A) y frontal (B).
Los estudios imagenológicos por ultrasonografía transfontanelar y ecografía abdominal revelaron un cerebro, así como unos órganos internos normales. De manera similar fue observado un tracto espinal normal a través del uso de la resonancia magnética (RM). Sin embargo, por ecocardiograma se identificó una restricción de la vena cava inferior (VCI).
Las radiografías comparativas de manos demostraron la ausencia del cuarto dígito, con fusión de la base del cuarto y quinto metacarpiano. Un engrosamiento y acotamiento del primero, segundo, tercero y quinto dígito, ausencia de las falanges medias en el segundo y tercer dígito, y artrodesis en las falanges proximal y media del quinto dedo de la mano derecha. La mano izquierda tuvo una aparente fusión del tercer y cuarto dígito. No hubo evidencia de compromiso en los huesos del carpo, ni de las epífisis distales del radio y el cúbito. La radiografía comparativa de pies puso en evidencia una tendencia a la polidactilia.
La tomografía computarizada de senos paranasales indicó el subdesarrollo de los senos paranasales, un puente nasal amplio, hipertelorismo, una reducción profunda de la órbita óptica con apariencia normal de los nervios y músculos ópticos (Fig. 4B). La historia clínica indicó que a los 21 días de edad el paciente fue hospitalizado debido a que presentó un episodio de taquipnea transitoria. A través del análisis de los exones 8, 9 y 10, encontramos una alteración en la secuencia del exón 8, la mutación S252W, previamente descrita en pacientes con SA (Fig. 2A).
Familia 2
Compuesta por tres personas, los padres sanos (I1 y II2) y el caso índice de SA (II 1) (Figura 1B).
II, 1: Paciente quien fue diagnosticado con SA al momento del nacimiento debido a la presencia de turricefalia, braquicefalia, frente prominente, hipoplasia medio-facial, hipertelorismo, puente nasal deprimido, hipoplasia malar y paladar alto. Ambas manos mostraron sindactilia ósea de los dígitos 1 al 5, excluyendo pulgares. Sindáctila ósea bilateral de todos los cinco dígitos sin presencia de falange distal. El análisis molecular identificó la variante T78.501A en la secuencia, la cual se encuentra cerca al lugar de hibridación de uno de los primers (Fig. 2B).
Discusión
El síndrome Apert es una Craneosinostosis Sindrómica, resultado de cambios en la secuencia del ADN, estas mutaciones podrían ocurrir en diferentes genes, uno de ellos el FGF y sus receptores, especialmente FGFR2, siendo P253R y S252W las mutaciones más frecuentes. Desde 1996, Slaney et al., relacionaron el LPH en pacientes con SA, especialmente a aquellos con la mutación S252W. La relación del LPH con esta mutación podría deberse a que estas patologías comparten vías de señalización 5. De igual manera, la formación de LPH de tipo no sindrómico ha sido asociada con las vías de señalización de FGF y FGFR, en donde las mutaciones de tipo Missense, tipo D138N y R84S en FGFR2 podrían alterar la función de la proteína debido al cambio en la afinidad del receptor 6.
A demás, es posible que el origen de las características fenotípicas en los dos pacientes afectados de la familia 1 (afección ósea y anormalidades craneofaciales), pueda deberse a la misma mutación en el gen FGFR2. No obstante, el análisis molecular identificó en el paciente II,2 la mutación S252W. Esta mutación incrementa la afinidad del receptor de manera inespecífica al subsecuente ligando FGF, permitiendo de este modo una activación inapropiada del receptor por ganancia de función, lo cual explicaría el fenotipo del paciente 7. En cuanto al paciente II, 1 con LPH no se encontró la misma mutación u otra ubicada dentro de los exones 8, 9 y 10, en los cuales la secuencia fue idéntica a la de su madre, quien no presenta características fenotípicas ni de LPH ni de SA. Estos resultados claramente indican que las características fenotípicas de ambos pacientes afectados podrían ser de orígenes diferentes.
No es posible descartar el origen de la mutación en el paciente II, 1 con fenotipo de LPH. Dado que el LPH aislado, es una alteración congénita compleja de origen multifactorial, en la mayoría de los casos, está causado por la interacción entre el medio ambiente y varios genes involucrados con la vía de señalización FGF, incluyendo FGFR1, FGFR2, y FGFR 3, FGF2, FGF3, FGF4, FGF7, FGF8, FGF9, FGF10 y FGF18, NUDT6, FCTBR, y PAX3 8, entre otros que podrían estar relacionados con esta vía de señalización. Se requieren estudios adicionales para determinar si hay mutaciones en alguno de estos que explique el fenotipo del paciente.
En la familia 2, identificamos en el paciente II, 1 una mutación en el ADN cerca de la región flanqueante del exón 8, a 20 pb del inicio del intrón. Aunque este tipo de variantes no ha sido descrita en humanos, estudios con modelos animales han demostrado una ganancia de función en la proteína codificada por el gen FGFR2, esto por la modificación de la secuencia intrónica que consecuentemente altera el sitio de splicing del exón. Por consiguiente, el fenotipo del paciente podría ser el resultado de la variante en la región intrónica del exón 8, la cual podría afectar el mecanismo del splicing y con esto la función del receptor, tal y como ha sido descrito en modelos animales 9.
Análisis adicionales del mARN del gen FGFR2 o directamente en el receptor podrían ayudar a esclarecer si la alteración en el sitio del splicing podría influenciar en la ganancia de función de la proteína 9.
Conclusiones
El SA y el LPH son dos patologías diferentes que comparten algunas vías de señalización relacionadas con el FGF y sus receptores, así como muchas otras proteínas, por lo cual es posible que un cambio en la secuencia del ADN de los genes involucrados resulte en la expresión de cualquiera de estos fenotipos. Sin embargo es necesario realizar estudios complementarios que incluyan más genes, permitiendo esto una visión más amplia para determinar la presencia o no de mutaciones que expliquen los fenotipos.
La mutaciones intrónicas podrían tener un efecto en el sitio de splicing y por ende en la función de la proteína, lo cual podría explicar el fenotipo del paciente II, 1 en la familia 2. Sin embargo para poner a prueba esta teoría es necesario llevar a cabo estudios funcionales.
Estos hallazgos son relevantes debido a que complementan el conocimiento actual del SA, permitiendo establecer nuevos cuestionamientos sobre las bases moleculares y vías asociadas al SA, así como de patologías asociadas como el LPH.