Introducción
La enfermedad de Charcot Marie Tooth (CMT), también conocida como Neuropatía Hereditaria Motora y Sensitiva (NHMS), es un grupo de desórdenes que se caracterizan por una polineuropatía motora y sensitiva crónica. Es la causa más frecuente de neuropatía periférica hereditaria y la patología hereditaria más común del sistema nervioso. Se presenta una prevalencia de 1 en 2,500 individuos 1. No hay diferencias en la prevalencia de la enfermedad en género o etnia y la edad de presentación es variable, dependiendo del tipo de CMT.
Las neuropatías hereditarias se clasifican en varias categorías con base en el tipo de herencia, edad de inicio y características de la conducción nerviosa 2. La enfermedad de Charcot Marie Tooth 1 (NHMS tipo I de acuerdo con la clasificación de Dyck), agrupa las neuropatías periféricas desmielinizantes, y las CMT2 (NHMS tipo II) comprenden las neuropatías periféricas axonales 3. Ambos grupos tienen un patrón de herencia autosómico dominante, que es el modo de herencia más frecuente en la enfermedad de CMT 4. La enfermedad de Charcot Marie Tooth 4 (NHMS autosómico recesivo) se refiere a las neuropatías con un patrón de herencia autosómico recesivo, e incluye tanto el tipo desmielinizante como el tipo axonal.La enfermedad de Charcot Marie Tooth 4A es el subtipo más común de CMT4 2. El único gen asociado con CMT4A es el gen GDAP1 2 , 4, que codifica para la proteína 1 asociada a la diferenciación inducida por gangliósido, una glutatión transferasa.
Se han descrito más de 40 mutaciones causantes de enfermedad en el gen GDAP1, tanto autosómicas dominantes como autosómicas recesivas 1, y la mutación Q163X es una de las más frecuentes. Esta mutación ha sido descrita como mutación fundadora en pacientes españoles, y se ha rastrado su origen hasta las regiones de Castilla, León y el País Vasco en España 5.
Presentamos el caso de dos pacientes de sexo femenino, hijas de padres consanguíneos, quienes presentaron una neuropatía axonal con inicio temprano de síntomas, y en quienes se encontró una mutación Q163X del gen GDAP1. Este es el primer caso de CMT autosómico recesivo reportado en nuestro país, así como el primer caso reportado de una mutación en el gen GDAP1.
Hemos hecho una revisión de la literatura, en donde describimos las características del gen GDAP1, particularmente la mutación Q163X y sus implicaciones en el desarrollo, diagnóstico y pronóstico del CMT4A.
Descripción del caso
Paciente 1
Se recibió en nuestra consulta una paciente femenina de 14 años, con un cuadro de deformidad progresiva en valgo de los miembros inferiores e hipotrofia muscular (Fig. 1a). Los síntomas se iniciaron a los 15 meses de edad. Posteriormente, desarrolló, de forma gradual, inestabilidad en la marcha, disminución de los arcos de movimiento en las manos y los brazos y un deterioro de las habilidades motoras gruesas y finas. Ella es el fruto de la segunda gestación de una madre Grávida 3 Para 3, y padres consanguíneos sanos. El embarazo fue normal, parto vaginal a las 39 semanas, con peso al nacer de 2,600 g (percentil 5), y una talla al nacer de 45 cm (percentil 25). Los hitos del desarrollo fueron normales, con sedestación a los 6 meses y marcha a los 15 meses de edad. La inteligencia es normal, con un desempeño académico normal. En la valoración inicial, encontramos una paciente de aspecto aparentemente sano, en silla de ruedas. El peso fue de 25 kg (percentil 3), talla de 132 cm (por debajo del percentil 3), y el perímetro cefálico fue de 52 cm (percentil 16). Los hallazgos al examen físico fueron: una disminución de la sensibilidad en las manos, los antebrazos, los pies y las pantorrillas, presentó escoliosis y pie cavo. La atrofia muscular fue evidente, con predominio en los antebrazos y las pantorrillas, y una evidencia de contractura de los dedos. No hubo una evidencia de dismorfia y fue llamativa la ausencia de parálisis de las cuerdas vocales o una dificultad respiratoria.
Figura 1.
A. Paciente Caso 1. B. Paciente Caso 2. Ambas pacientes desarrollaron síntomas similares. Nótese la hipotrofia muscular simétrica y severa de las extremidades, la contractura y deformidad de los dedos y codos, la marcada atrofia de los músculos tenares, hipotenares e interóseos de manos, el pie cavo, y la ausencia de dismorfia.
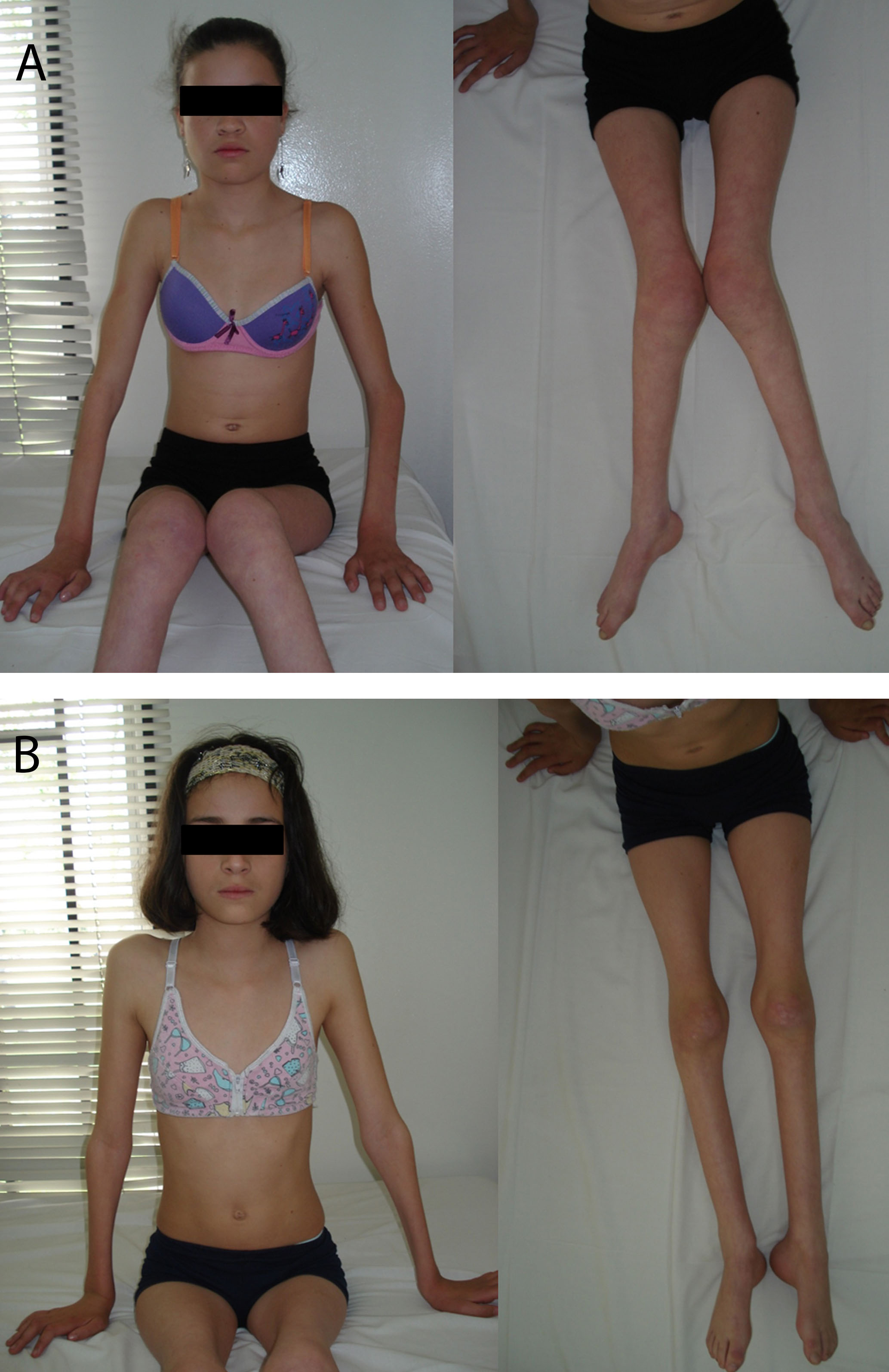
Presentó además, una disminución de los reflejos aquiliano y bicipital; la fuerza de los miembros superiores fue de 4/5 en las manos, 4/5 en los antebrazos y 4/5 en la cintura escapular (de acuerdo con la escala MRC). Los miembros inferiores presentaron una fuerza de 3/5 en los muslos, las pantorrillas y los pies. Los nervios craneales eran sanos, y no se encontraron signos de disautonomía.
La resonancia magnética cerebral fue normal, así como las enzimas hepáticas. Las velocidades de neuroconducción motora y sensitiva (NCV) estuvieron ausentes en todos los nervios explorados. La electromiografía (EMG) de las extremidades inferiores mostraron actividad insercional normal en todos los músculos explorados; hubo actividad espontánea anormal en el músculo recto femoral derecho; se evidenció un aumento de la amplitud y reclutamiento reducido en los músculos tibial anterior izquierdo, gastrocnemio izquierdo y recto femoral izquierdo.En conclusión, la EMG y la NCV mostraron una neuropatía sensitiva y motora de tipo axonal, con signos de re inervación, que es consistente con un proceso crónico de manifestación severa.
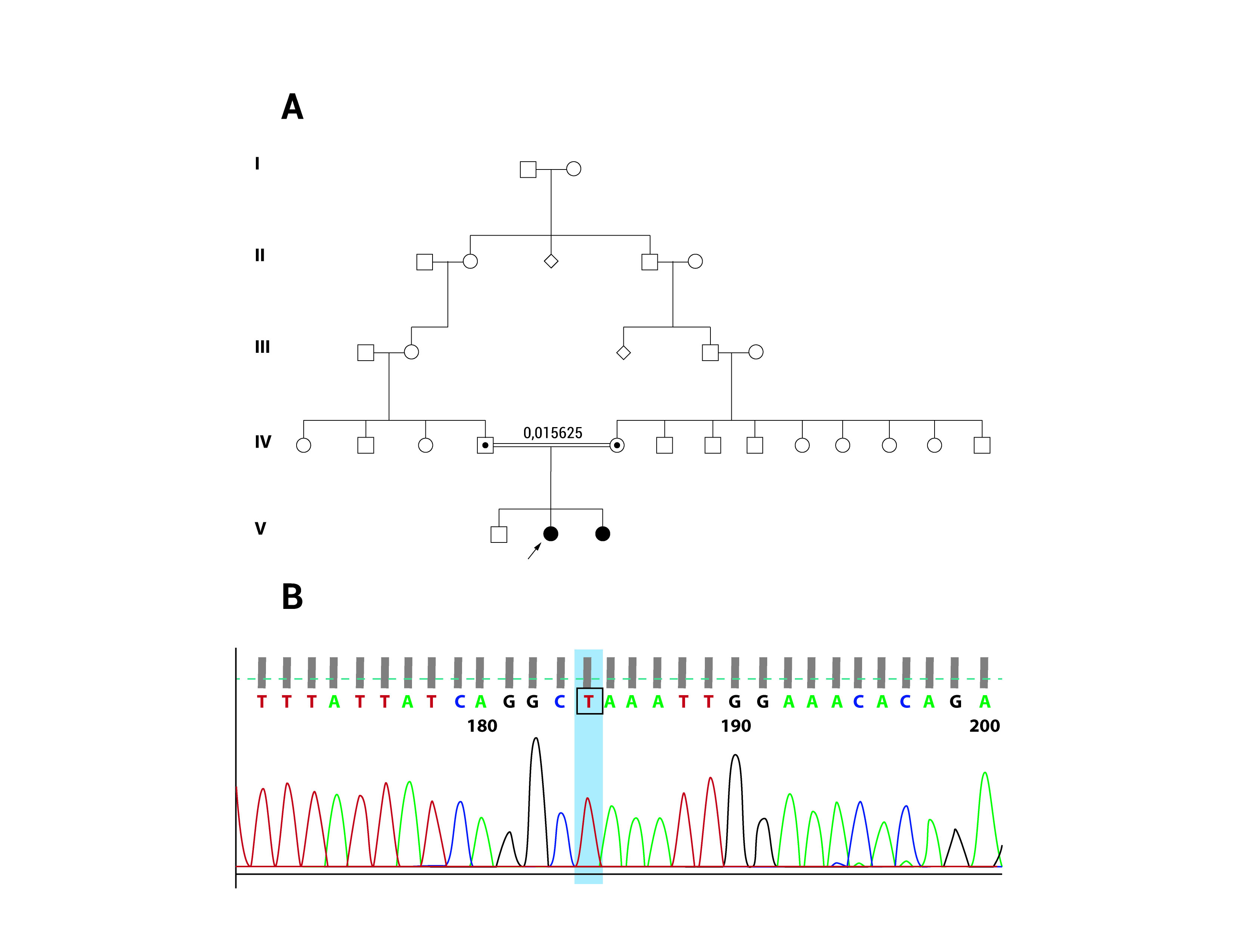
Se solicitó un estudio de secuenciación del gen GDAP1 en esta paciente. El estudio identificó una mutación puntual homocigota (c.487 C>T) en el exón 4, dando como resultado un codón de parada prematuro (p.Q163X) (Fig. 2). Este resultado confirmó el diagnóstico de enfermedad de Charcot Marie Tooth tipo 4A (autosómica recesiva, tipo axonal).
Figura 2.
Arbol familiar de los pacientes. A.La flecha señala la paciente Caso 1. El hermano mayor no está afectado, sin embargo, no se realizó prueba genética por la edad del paciente. No hay otros miembros afectados conocidos en la familia, y no se dispone del estado de portador para otros miembros de la familia. B. Resultados de la secuenciación del gen GDAP1 de la paciente Caso 1, que muestra una mutación Q163X (c.487 C>T).
Paciente 2
Esta paciente de 10 años, quien es la hermana menor de la primera paciente descrita anteriormente, fue referida a nuestro servicio por presentar deformidad progresiva en valgo de los miembros inferiores, alteración de la marcha y caídas múltiples (Fig. 1b). Los síntomas se iniciaron a los 15 meses de edad. A los 3 años desarrolló hipotrofia muscular progresiva y una disminución de los arcos de movilidad en las manos a los 4 años de edad. Ella es el fruto de la tercera gestación de padres consanguíneos. El embarazo fue normal, parto vaginal a las 40 semanas, con peso al nacer de 2,600 g (percentil 5), talla al nacer de 46 cm (percentil 25-50) y perímetro cefálico de 46 cm (percentil 25-50). Presentó ictericia neonatal, manejada con fototerapia. Los hitos del desarrollo fueron normales, con sedestación a los 7 meses, y marcha a los 12 meses de edad. A la inspección, encontramos una paciente en muletas, pero quien conserva la deambulación. Por otro lado, es una niña aparentemente sana. El peso fue de 22 kg (percentil 5), la talla fue de 129 cm (entre los percentiles 10 y 25) y el perímetro cefálico fue de 53 cm (percentil 50).
Al examen físico, se evidenció atrofia muscular en las regiones tenar e hipotenar de las palmas y en los músculos interóseos de ambas manos, así como atrofia muscular en miembros inferiores y presentó pie cavo. Los reflejos aquiliano y bicipital estaban ausentes. La fuerza fue de 3/5 en los pies, las pantorrillas y los muslos y de 4/5 en las manos, los antebrazos y la cintura escapular (de acuerdo con la escala MRC). No se encontró dismorfia, tampoco parálisis de las cuerdas vocales, ni dificultad respiratoria.
Las velocidades de neuroconducción estuvieron ausentes en todos los nervios explorados. La EMG de los miembros superiores e inferiores mostró actividad insercional normal en todos los músculos explorados, sin actividad espontánea anormal. Se evidenció una disminución del reclutamiento en todos los músculos explorados, y se encontró un aumento de la amplitud con morfología polifásica en los músculos tibial anterior derecho y bíceps femoral izquierdo. En conclusión, la EMG y NCV mostraron una neuropatía sensitiva y motora de tipo axonal, con signos de re inervación.
Discusión
La revisión hecha por Yiu y Ryan 6 brinda una metodología útil para el enfoque diagnóstico del niño con neuropatía. A diferencia del adulto, en quien la principal causa de neuropatía es la diabetes, en los niños es la enfermedad genética. Las características de la neuropatía de origen genético son el retraso de los hitos del desarrollo motor (cuando se desarrolla en el primer año de vida), debilidad proximal y deformidad en los pies o escoliosis. En estos niños se debe evaluar si la neuropatía se asocia con un desorden del sistema nervioso central, si la neuropatía es axonal o desmielinizante, el patrón de herencia, y otras características particulares (dismorfía, retardo mental, otras manifestaciones neuropsicológicas).
Las pacientes de este caso tuvieron un inicio de la enfermedad en los primeros dos años de vida, con hipotrofia progresiva y simétrica, inestabilidad de la marcha y deformidad de pies, en el momento en que ya habían completado los hitos del desarrollo de acuerdo con su edad. Ambas mostraron un desarrollo similar de la enfermedad, y ninguna de las dos presentó dismorfia o retardo mental. Estos hallazgos, en el contexto de una neuropatía, guiaron a los autores al diagnóstico de una neuropatía hereditaria sensitiva y motora.
El EMG y el NCV mostraron una neuropatía axonal severa en ambas pacientes, lo que sugiere el diagnostico de enfermedad de CMT. Hicimos una revisión del árbol genealógico de las pacientes (Fig. 2). Los padres son primos en primer grado, ambos sin antecedentes médicos de importancia y no hay otros miembros afectados en la familia. Las pacientes tienen un hermano mayor sano. El patrón de herencia mostrado en el árbol genealógico sugiere una enfermedad autosómica recesiva 7.
Nosotros seguimos el algoritmo elaborado por England et al.8, sobre la base de una historia familiar relevante (dos miembros afectados), dos pacientes con una neuropatía axonal, la edad de inicio, y un patrón de herencia autosómico recesivo. Por tanto, se solicitó la secuenciación del gen GDAP1 en la primera paciente.La secuenciación del gen GDAP1 mostro una mutación puntual homocigota (p.Q163X), lo que nos llevó al diagnóstico de enfermedad de CMT, tipo 4A. En consecuencia, no se solicitó biopsia de nervio. La biopsia de nervio se reserva para aquellos pacientes en quienes las pruebas genéticas no arrojan un diagnóstico molecular, en quienes la presentación de la enfermedad es atípica, o en quienes se sospecha neuropatía inflamatoria 9. No se encontró ninguna de estas características en nuestras pacientes.
El modo de herencia es autosómico recesivo. Ambos padres son portadores obligados de la mutación y no tienen riesgo de desarrollar la enfermedad. Los hermanos de los padres tienen un 50% de riesgo de ser portadores. Después de obtenido el resultado genético, se realizó asesoramiento genético a los padres acerca de la enfermedad.
El hermano mayor de las pacientes fue descrito como normal por sus padres. Los hitos del desarrollo fueron normales, y no mostró signos de neuropatía o retardo mental. El fenotipo y el examen físico fueron normales. En este caso, consideramos que el hermano no está afectado y como hermano de un individuo afectado, tiene una probabilidad de 2/3 de ser portador. La decisión de realizarse el examen genético debe ser hecha por el mismo paciente, cuando alcance la mayoría de edad. En el caso de ser portador, se debe ofrecer asesoramiento genético, así como pruebas prenatales o preimplantacionales, con el fin de discutir las opciones reproductivas y el riesgo de los posibles hijos.
Características de la proteína GDAP1
El locus del gen GDAP1 se encuentra en el cromosoma 8q13-q21. La proteína se localiza en la membrana mitocondrial externa 1 como una proteína integral de membrana10. Se encuentra principalmente en las células neuronales del sistema nervioso central y periférico1, pero también se expresa en las células mielínicas de Schwann del sistema nervioso periférico 10. La proteína GDAP1 aumenta la enzima glutatión-s-transferasa celular en las neuronas, disminuyendo la producción de especies reactivas de oxígeno, y protegiendo las células del estrés oxidativo 1. La proteína GDAP1 también tiene una función en el balance entre las mitocondrias fusionadas y fragmentadas 10, lo que parece ser esencial para la integridad de los nervios periféricos mielínicos, y es también un factor de fisión peroxisomal, la ausencia de GDAP1 causa una alteración de la morfología del peroxisoma y su sobreexpresión promueve la fragmentación peroxisomal 11.
Se han identificado más de 40 mutaciones causales de la enfermedad en el gen GDAP1 1. Pueden encontrarse patrones desmielinizantes (R161H), axonales (S194X) e intermedios (T117fs) en el NCV, así como patrones de herencia autosómico dominante (R120W, T157P, Q218E, C240Y) 12 o recesivo (Q163X); puede encontrarse un amplio espectro de características clínicas y el curso de la enfermedad es diferente entre cada tipo de mutación del gen GDAP1. Las mutaciones dominantes del gen GDAP1 se asocian con neuropatía axonal leve de inicio tardío y progresión lenta; la extensión y severidad de la debilidad proximal es menos pronunciada comparada con las mutaciones recesivas, y los hallazgos electrofisiológicos son variables 12,13. Las mutaciones recesivas del gen GDAP1 tienen un fenotipo más severo y un inicio más temprano, por lo general antes de los 10 años; la parálisis de cuerdas vocales, la debilidad facial y la parálisis diafragmática son hallazgos frecuentes 12.
La mutación Q163X (c.487 C>T) es una de las más comunes del gen GDAP1 12. Esta mutación se localiza en el exón 4, y da como resultado un codón de parada prematuro que afecta el dominio GST 14. Niemann et al.10, mostraron que la proteína GDAP1 mutada no es detectable en células transfectadas, lo que sugiere que la proteína es altamente inestable, y que la mutación se comporta como una mutación nula.
Cuesta et al. 15, reportaron tres diferentes familias de ascendencia española, en las cuales se encontraba presente la mutación Q163X de forma homocigota, o como heterocigoto compuesto con la mutación S194X. Boerkoel et al.16, reportaron cinco familias hispanas con mutación homocigota Q163X, cuatro procedentes de Costa Rica, y una procedente de Perú.
Claramunt et al.5, demostraron que la mutación Q163X es la más frecuente en España, con una edad estimada de 33,000 años (1,650 generaciones, 20 años por generación). Ellos hicieron una comparación entre los haplotipos de los pacientes con la mutación Q163X provenientes de Castilla, León, el País Vasco y la región de Valencia en los pacientes reportados por Cuesta et al.15, y los haplotipos de los pacientes descritos por Boerkoel et al.16, y confirmaron que las mutaciones encontradas en estos pacientes tienen un origen ancestral común. Esto sugiere la existencia de un efecto fundador. Anteriormente, no se ha descrito esta mutación en Colombia. Es posible que esta mutación tenga su origen en la misma mutación fundadora descrita previamente, sin embargo, se requieren más estudios para confirmar esta hipótesis.
Si bien los pacientes con una mutación Q163X tienen fenotipos heterogéneos, aún dentro de la misma familia, comparten algunas características que diferencian el CMT4A causado por la mutación en GDAP1 Q163X, de otras formas de CMT. Las características fenotípicas principales descritas 15 de los pacientes con la mutación GDAP1 Q163X, son el inicio temprano en la niñez de debilidad e hipotrofia en las manos y los pies, estudios electrofisiológicos con patrón axonal, disfonía y parálisis de las cuerdas vocales de inicio en la segunda década de la vida, e inteligencia normal. Se puede encontrar también un pie cavo, manos en forma de garra y contracturas musculares. La mayoría de estos pacientes se encuentran en silla de ruedas después de los 10 años de edad.
Otros autores 16,17 han descrito un fenotipo definido por el inicio temprano de los síntomas, por lo general durante el primer año de vida, que consiste en una debilidad muscular distal progresiva, una pérdida de la sensibilidad, marcha en estepaje, pie cavo, cifoescoliosis, dedos en posición de martillo, y reflejos disminuidos o ausentes. Los pacientes se encuentran en silla de ruedas alrededor de la segunda década de la vida. Los hitos del desarrollo se alcanzan a una edad apropiada previa al desarrollo de la enfermedad, y la inteligencia se preserva.
Los pacientes presentados en este reporte de caso muestran prácticamente todas las características descritas en estos reportes de casos. No se encontró disfonía, parálisis de cuerdas vocales ni parálisis diafragmática al momento de la publicación de este artículo. La espirometría fue normal en ambas pacientes, lo que sugiere no hay compromiso vocal o diafragmático.
La NCV y la EMG en los pacientes con mutación Q163X son heterogéneos. Boerkoel et al. 16, reportaron velocidades de neuroconducción motora del nervio mediano indetectables en 4 de 5 pacientes, y velocidades de neuroconducción sensitiva en 2 de 5 pacientes. Claramunt et al. 5, reportaron NCV entre 24 y 45 m/s. Los pacientes reportados por Sevilla et al. 17, tenían NCV por encima de los 40 m/s, y los pacientes sin reporte debido a la atrofia muscular distal severa, tenían conservadas las latencias motoras proximales. En ambas pacientes de nuestro reporte, el EMG reportó un patron axonal; por otro lado, la NCV estuvo ausente en todos los músculos explorados. Es posible que la causa de estos hallazgos sean el estadio avanzado de la enfermedad y la hipotrofia muscular severa.
Los hallazgos histopatológicos asociados con la mutación Q163X incluyen la pérdida severa de axones mielinizados, pérdida moderada de fibras amielínicas, y formaciones en bulbo de cebolla, sin evidencia de renovación axonal 16. La formación de bulbos de cebolla es menos evidente en comparación con pacientes heterocigotos compuestos 17.
Conclusión
El estudio de la genealogía del paciente, las características fenotípicas, y los hallazgos de la NCV y EMG deben incluirse como herramientas valiosas en el enfoque clínico del paciente con enfermedad de CMT tipo axonal, autosómica recesiva. Se debe considerar la presencia de mutaciones del gen GDAP1 en pacientes de origen suramericano, y principalmente la mutación Q163X, como causa de CMT4A. Se necesita mayor información del transfondo genético de la enfermedad CMT4A en Colombia y otros países suramericanos, con el fin de describir y entender su curso y pronóstico clínico.