Introducción
La enfermedad cerebrovascular (ECV) es un problema de salud pública en el mundo. Corresponde a la principal causa de incapacidad permanente en adultos y la tercera causa de muerte en los países desarrollados 1. Una de cada seis personas sufre accidentes cerebrovasculares (ACV) consecuencia de la ECV durante su vida y cerca del 30% presentan disfunciones graves o permanentes que los hacen dependientes de otras personas. De acuerdo al territorio cerebral comprometido, las alteraciones pueden ser motoras, sensoriales, mnésicas o cognitivas, entre otras, aunque por lo general el compromiso es mixto 2. Las alteraciones pueden modificarse con el tiempo debido a cambios exofocales ocasionados desde la lesión por un proceso de plasticidad del sistema nervioso denominado diásquisis 3-5.
Los ACV se pueden dividir en isquémicos, por disminución del flujo sanguíneo en determinada área cerebral; o hemorrágicos, por rotura arterial intracraneal. La práctica clínica da cuenta de que el 85% de los infartos cerebrales son isquémicos; uno de los más comunes es la isquemia focal por obstrucción de la arteria cerebral media (ACM) 6.
La isquemia cerebral es un proceso fisiopatológico caracterizado por la disminución transitoria o permanente, focal o global, del flujo sanguíneo en el encéfalo que causa diferentes patrones de disfunción del tejido de acuerdo a la severidad y la duración de la isquemia 7. Cuando el flujo de oxígeno y glucosa está por debajo de 10 mL/100 g/min, hay pérdida de la función de la membrana celular por agotamiento metabólico de las bombas de ATP, se pierde el potencial iónico y se presenta edema celular por incremento de sodio intracelular; conduciendo en minutos a la necrosis del tejido y al desencadenamiento de procesos inflamatorios 8. Asimismo, hay un aumento de la actividad eléctrica de las membranas celulares al borde del foco; debido al aumento de potasio y glutamato extracelular. Esto aumenta la excitabilidad en las células, incrementa el gasto metabólico y lleva al crecimiento del área infartada. El área directamente afectada se denomina foco isquémico y es la región que muestra las primeras consecuencias de la lesión 9-10.
Adyacente al foco isquémico se establece la penumbra, un área con poca perfusión en la que persiste un flujo residual, capaz de mantener con vida a las neuronas. Sin embargo, éstas son disfuncionales debido al estrés metabólico. Como en la penumbra no se ha establecido la necrosis, es un área susceptible de recuperación; no obstante, si no se presenta una adecuada reperfusión se presenta necrosis y estabilización del déficit clínico 7,11. La penumbra es uno de los objetivos terapéuticos para reducir las secuelas de la isquemia, dado que allí se desencadenan procesos de angiogénesis necesarios para restablecer el flujo sanguíneo y al parecer, para estimular la neurogénesis endógena 12. No obstante, hay sectores del área de penumbra que no se restauran y que a pesar de la normalización del flujo sanguíneo, sufren muerte celular apoptótica días o meses después de la isquemia 13.
La cascada isquémica no solamente afecta el foco y el área de penumbra, sino también áreas exofocales, ipsilaterales y contralaterales a la lesión, además estructuras como el cerebelo, el tálamo, la sustancia negra y el hipocampo.. Estas áreas y estructuras se alteran en virtud de sus conexiones anatómicas o funcionales con el foco isquémico y con el área de penumbra 14-16. Este fenómeno no sólo se relaciona con procesos patológicos, sino con la reorganización del sistema nervioso y recuperación funcional 17 y se observa en otros trastornos como tumores, epilepsia y lesiones traumáticas 18.
Hay evidencia de diásquisis transcallosa o contralateral debido al hallazgo de afecciones cerebrales bilaterales tras lesiones unilaterales. Teniendo como estímulo la lesión, la reorganización del hemisferio no afectado se presenta gracias a la desinhibición de vías comisurales y al relativo incremento de la excitabilidad neuronal subcortical, especialmente talámica, probablemente como un recurso para contrarrestar la pérdida de función contralateral 19,20. En este sentido, las alteraciones exofocales contralaterales se pueden asociar con plasticidad cerebral, mecanismos de reorganización neuronal y procesos compensatorios y adaptativos 21-25. Se ha identificado que si se realiza ablación del cuerpo calloso los efectos contralaterales se reducen o no se presentan y que estos cambios generan tolerancia a futuros eventos isquémicos 24, 26.
En este aspecto, se ha descrito la activación de más de 80 genes en la corteza cerebral ipsilateral y contralateral, en el hipocampo, estriado y tálamo, que se relacionan con modificaciones circulatorias y metabólicas 27. También se ha demostrado arborización dendrítica y crecimiento axonal en el hemisferio contralateral, expresión de neurotrofinas con recuperación motora y sensorial, relacionados con la organización del sistema piramidal después de una isquemia 28. Además, hay evidencia de procesos de sinaptogénesis y neurogénesis en algunas áreas y cambios adaptativos en la corteza entorrinal, entre otros 24,25,29.
Aunque no es claro el mecanismo de la diásquisis transcortical 30, clínicamente, se sabe que las alteraciones contralaterales contribuyen al déficit neurológico y a la heterogeneidad de signos y síntomas a mediano y largo plazo como déficit cognitivo, pérdida de memoria motora, afasia y amnesia episódica 31,32. No obstante, se conocen pocas propuestas para tratar este aspecto del daño cerebral 2-4. De lo anterior se concluye que es importante el estudio del hemisferio contralateral tras la lesión cerebral isquémica como aporte a las teorías de la recuperación y reorganización de las funciones cerebrales. Esto reviste interés clínico debido a que en muchas ocasiones la disfunción y la recuperación no se relacionan directamente con el área y tamaño de la lesión 33. Conocer la fisiopatología de la isquemia y los cambios transcorticales son aspectos fundamentales para entender los procesos de restauración de los sistemas afectados por la lesión y, consecuentemente. poder determinar el tipo de tratamiento más eficaz.
El objetivo de esta investigación fue la caracterización descriptiva de los cambios corticales contralaterales en un modelo experimental de isquemia cerebral focal para evidenciar procesos de reorganización celular, con el fin de entender los cambios fisiopatológicos. Se utilizó el anticuerpo monoclonal específico para neuronas anti-NeuN (Neuronal Nuclei - MAB377) con el cual además del núcleo se logra marcar el soma y los procesos proximales de las neuronas. La marcación no incluye las células gliales ni los componentes vasculares 34, a diferencia de los métodos histológicos convencionales que eventualmente pueden causar "ruido" en las observaciones de las células. Dentro del núcleo, la reactividad al NeuN se encuentra preferentemente en las zonas donde hay baja densidad de cromatina, y prácticamente se excluye de las áreas que contienen ADN densamente empaquetado. La desfosforilación enzimática suprime la inmunoreactividad al NeuN, confirmando que éste es de hecho una fosfoproteína, y se establece que la unión al anticuerpo depende del estado de fosforilación 9. Es claro que las circunstancias de fosforilación de proteínas se alteran significativamente en la isquemia. El NeuN se ha identificado con la proteína Fox-3 que, como otras proteínas de este tipo, regula el empalme del mRNA 35.
En circunstancias patológicas como la isquemia cerebral, la disminución en la cuantificación de células NeuN puede indicar pérdida neuronal por necrosis 34. Adicionalmente se ha descrito una correlación entre células apoptóticas, túnel positivo e inmunoreactividad al NeuN, colocalización que relaciona la disminución de la inmunoreactividad al NeuN con muerte celular apoptótica 36. Sin embargo, se ha establecido que la disminución en la inmunoreactividad al NeuN puede estar relacionada con la depleción de proteína o por disminución de la inmunoreactividad; cambios no necesariamente relacionados con muerte celular, pero sí con estrés metabólico celular 37. Si consideramos que la diásquisis transcallosa corresponde a un fenómeno de estrés celular, evaluar la inmunoreactividad del NeuN en la corteza contralateral a la lesión puede constituirse en un sensible marcador de fenómenos exofocales.
Materiales y Métodos
Se intervinieron 28 ratas machos Wistar adultas con un peso entre 240 y 320 g (promedio 280 g), con edad entre 120 y 180 días; siguiendo el protocolo para evitar el sufrimiento innecesario.
Procedimiento microquirúrgico
Después de un período de 6 h de ayuno los especímenes experimentales fueron anestesiados con clorhidrato de ketamina (40 mg/kg) y atropina (0.1 mg/kg) vía intraperitoneal. La técnica de isquemia cerebral aplicada en este trabajo fue desarrollada por Zea et al. 38, y perfeccionada por Belayeb et al, 39. Bajo microscopio quirúrgico se realizó una incisión longitudinal en la línea media del cuello. Se localizaron y disecaron la arteria carótida común (ACC) y la arteria carótida externa (ACE), que se seccionó, quedando así libre la ACE y ligada en su extremo. Un segmento de nylon monofilamento 4-0 de 3.5 cm se introdujo por el extremo del fondo de saco correspondiente a la ACE hasta una longitud de 17.5 mm y se fijó. Al cabo de 90 min se hala el nylon a través de la herida quirúrgica y se dejó alojado su extremo en el fondo de saco de la ACE para iniciar la reperfusión. A los 4 especímenes del grupo control se les realizó todo el procedimiento descrito a excepción de que el nylon monofilamento 4-0 no se introdujo hasta el nivel de la ACM. Durante el procedimiento quirúrgico se evaluó regularmente la función cardiorrespiratoria y la temperatura rectal. Se mantuvo la temperatura a 37° C mediante una lámpara de calor. Se establecieron 5 grupos, uno control con 4 especímenes, y los cuatro restantes con sobrevida de 24 h, 96 h, 10 días y 20 días cada uno con 6 especímenes.
Inmunohistoquímica
Al tiempo correspondiente, los especímenes de cada grupo fueron anestesiados y perfundidos por vía transcardiaca a través del ventrículo izquierdo con 200 mL de solución salina al 0.9% seguido de 200 mL de una mezcla de paraformaldehido (4.0%), lisina y periodato de sodio en buffer fosfato 0.1 M (pH 7.4). Seguidamente los cerebros se retiraron de la caja craneal y se dejaron en el fijador. Después se practicaron 10 cortes coronales de 30 μm de espesor en el vibrátomo de medio líquido, los cortes se realizaron de acuerdo con las coordenadas del Atlas de Paxinos y Watson 40 de forma secuencial anteroposterior partiendo del punto interaural 10.7 con bregma 1.7, cada milímetro hasta el punto interaural 3.7 con bregma -5.3. Los cortes se incubaron con el anticuerpo primario NeuN (Neuronal Nuclei - MAB377 Chemicon) por 8 h, después se lavaron en buffer fosfato 10 mM (pH 7.4) y se hicieron reaccionar con el anticuerpo secundario utilizando el kit de avidina biotina (vectastain), para ser revelados utilizando diamino benzidina contrastado con níquel (ABC kit vector laboratories). Los cortes seleccionados de las muestras como control al proceso de inmunohistoquímica tuvieron el mismo procedimiento anteriormente descrito, excepto por la incubación con el anticuerpo primario.
Los cortes de los diferentes tiempos experimentes fueron teñidos con 2,3,5-Triphenyltetrazolium chloride (TTC) para observar el área infartada, para este tipo de tinción vital uno de las ratas de cada uno de los grupos no fue perfundida, se anestesió profundamente y se decapito, se retiró rápidamente el cerebro de la caja craneal. Los cerebros fueron cortados coronalmente cada 2 mm a partir del polo frontal utilizando una matriz cerebral (Stoelting). Las secciones cerebrales fueron sumergidas en una solución amortiguadora de fosfato salino (PBS) 0.1 M pH 7.2 que contenía TTC al 0.5% por 30 min a 37° C. Finalmente, las secciones teñidas fueron fijadas en formalina al 4% y mantenidas a 4° C. Seis secciones por cada cerebro fueron fotografiadas en sus aspectos anterior y posterior y las áreas isquémicas fueron medidas manualmente de manera ciega. El análisis morfométrico y volumétrico implicó corregir el error ocasionado por el edema, para lo cual se utilizó la fórmula [(área del hemisferio normal / área del hemisferio infartado) x área del infarto] 41. Todos los especímenes experimentales presentaron lesión en un área proporcional al corte coronal aproximado en Bregma 2.04 mm de 39.4 ± 4.2%. No hubo diferencias mayores entre los especímenes y la variabilidad en el tamaño entre los grupos fue muy baja. El volumen del daño isquémico se calculó integrando el área de la lesión corregida de los diferentes cortes coronales con la coordenada anteroposterior 42, de esta manera el resultado se expresa en milímetros cúbicos de lesión, se observó una importante homogeneidad en los diferentes especímenes 162.5 ± 17.5 mm3.
La evaluación de los cortes procesados para NeuN por inmunohistoquimica se realizó de manera cualitativa semiestructurada puesto que cada uno de los investigadores examinó de manera independiente las placas a 10x y a 40x con los cortes de los especímenes del grupo control y de los experimentales, centrando la atención en cortes correspondientes a corteza frontoparietal, esto se hizo de manera sistemática siguiendo una secuencia codificada con un orden previamente establecido y sin conocimiento del tipo de espécimen que se encontraba observando. Previo a este procedimiento se realizó una evaluación y entrenamiento de todos los investigadores para garantizar la mayor objetividad posible en las observaciones. Se inició con una observación global de las láminas a 10x y después una revisión lámina por lámina a 40x desde la I a la VI, prestando atención al grado de inmunoreactividad de las neuronas al NeuN (intensidad de la inmunoreactividad similar al control v/s intensidad de la inmunoreactividad menor que el control - morfología neuronal similar al control v/s morfología neuronal diferente al control). Cada una de las descripciones fueron registradas en el mismo tipo de formato para ser recopiladas por el investigador principal, se seleccionaron imágenes para el análisis de resultados cuando la coincidencia en la observación fue del 80% o más. Para obtener las imágenes se les solicitó a los investigadores seleccionar tres patrones consideradas por ellos como representativas de cada cambio observado como relevante, de esta selección a su vez el investigador principal tomo al azar las imágenes para ilustrar la publicación y para el análisis final.
Por otro lado se cuantificaron los cambios en el número de células NeuN positivas de la lámina VI: se demarcaron tres áreas de tamaño similar por cada uno de los especímenes, se fotografiaron y se procesaron en blanco y negro al 50% en el formato de imagen del programa World de Office, se imprimieron las imágenes y se contaron manualmente las neuronas. Cada uno de los investigadores envió los resultados y se integraron mediante una sumatoria simple para definir la proporción de neuronas con relación al control.
Ética
El control del estado de los 28 sujetos en todas las fases de la investigación se realizó utilizando un protocolo de supervisión que establece criterios para evitar el sufrimiento innecesario, especialmente después de la intervención quirúrgica. El protocolo experimental fue evaluado y autorizado por el comité de ética de la Universidad Libre de Cali, cumple la normativa vigente de la Comunidad Económica Europea para el uso y cuidado de los animales utilizados para la experimentación y otras finalidades científicas (Strasbourg, 15 junio del 2006) y de la legislación colombiana (Ley 84 de 1989 y Resolución No. 8430 de 1993 del Ministerio de Salud) sobre ética, cuidado y control de animales para fines experimentales.
Resultados
La mortalidad en las ratas fue del 21%, distribuida por grupos así: Control (0%), 24 h (7.1%), 96 h (3.6%), 10 días (3.6%) y 20 días (7.1%). Se procesaron del grupo control 6 especímenes, de los grupos experimentales de 24 h 4 especímenes, 96 h 5 especímenes, 10 días 5 especímenes y 20 días 4 especímenes.
En este estudio se evaluaron de manera cualitativa los cambios en la región contralateral a la lesión de la corteza cerebral frontoparietal. Los resultados se obtuvieron con base en los datos aportados por los grupos de ratas experimentales confrontados con el grupo control y el análisis descriptivo de los cambios en la inmunoreactividad de las neuronas al NeuN. En todos los especímenes sometidos a isquemia y reperfusión se observó una lesión frontoparietal en el territorio de la ACM del tejido ipsilateral a la lesión, lo que comprende una pérdida de las características anatómicas e histológicas con grados de daño neuronal que van desde lesión evidente a las 24 h, hasta pérdida total del tejido con formación de zonas quísticas (Fig. 1), como lo hemos reportado en otros estudios 4,9,33.
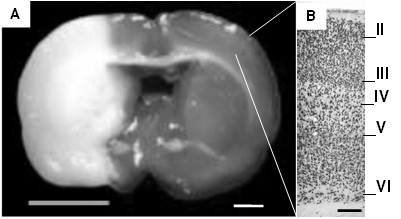
Figura 1.
Corte coronal teñido con TTC después de isquemia cerebral focal y reperfusión con la técnica de filamento intraluminal. En A, se observa pálida el área de lesión que incluye corteza cerebral y ganglios basales. A la derecha se observa la corteza cerebral ampliada a 10X en la que se diferencian las neuronas marcadas con NeuN organizadas en láminas. Las observaciones descriptivas de todos los especímenes se tomaros del sector aproximado señalado en la imagen que corresponde a un corte coronal en Bregma 2,04 mm. (A barra= 2 mm, B barra= 250 μm).
A las 24 h, en zona contralateral se presentaron cambios en las neuronas con disminución de la inmunoreactividad al NeuN en comparación con los controles (Fig. 2 C y D), estos cambios son notables en las láminas supragranulares. A las 96 h postisquemia, en la corteza del hemisferio contralateral a la lesión, es característica la diminución de la inmunoreactividad celular en capas supra e infragranulares. Además, se aprecian núcleos celulares con fraccionamiento y consolidación con apariencia morfológica que se asemeja a la fragmentación nuclear observada en la muerte celular apoptótica (Fig. 2 E y F). A las 96 h se aprecian áreas de transición de un tejido organizado a uno donde se pierde la apariencia citoarquitectónica especialmente en la capa VI, que incluye áreas de alteración en la continuidad de la lámina y una aparente pérdida neuronal (Fig. 3 C).
Figura 2.
Imágenes representativas de la inmunoreactividad al NeuN en corteza frontoparietal contralateral a la lesión isquémica en un modelo experimental de isquemia cerebral focal de la arteria cerebral media. A y B, imágenes control de la zona infragranular a nivel de la lámina V donde se observa una adecuada inmunoreactividad a las 24 h. C y D, disminución de la inmunoreactividad a las 24 h en corteza cerebral supragranular a nivel de la lámina III (flechas en D). En E y F, a las 96 h, se observa en corteza cerebral infragranular a nivel de la lámina V, neuronas y núcleos celulares con inmunoreactividad heterogénea (las flechas delgadas señalan neuronas con disminución de la inmunoreactividad, las flechas gruesas neuronas con núcleos fraccionados y consolidados). G y H, inmunoreactividad a los 10 días, supragranular a nivel de la lámina III similar al control. Las observaciones descriptivas de todos los especímenes corresponden a cortes coronales entre Bregma 1.80 mm a 2.16 mm, aproximación mediodorsal 3.85 a 4.8 mm y dorsoventral 2 a 3 mm. (Primera columna barra= 250 μm, segunda columna barra= 25 μm).
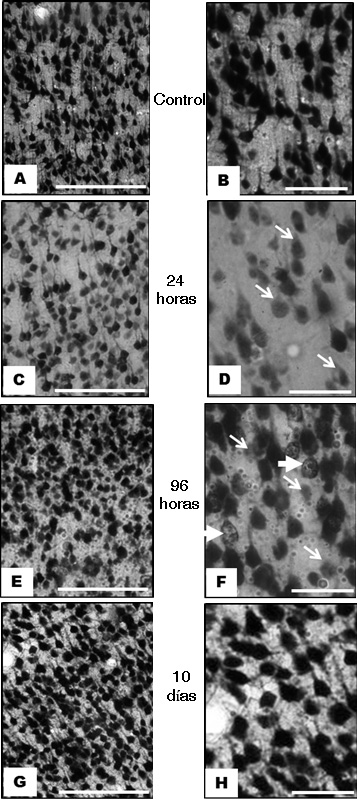
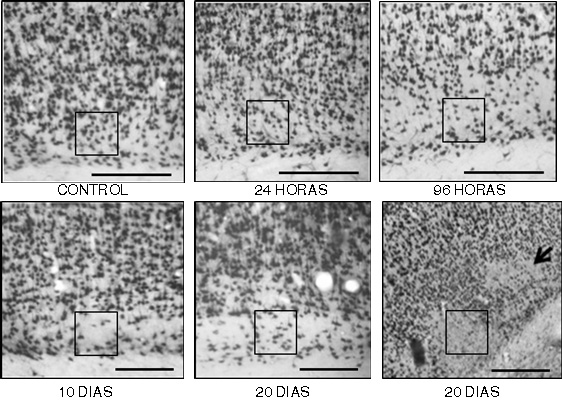
Figura 3.
Imágenes representativas de la inmunoreactividad al NeuN de la lámina VI en corteza frontoparietal contralateral a la lesión isquémica en un modelo experimental de isquemia cerebral focal de la arteria cerebral media. A control, B a F cambios en la lámina VI notable en el transcurso del tiempo, se señalan con los recuadros áreas con alteración en la inmunoreactividad al NeuN y aparente disminución de la celularidad, en F con vista panorámica a 10X de aumento, la pérdida se observa en parches en algunos sectores de la lámina VI. El número de neuronas inmunoreactivas disminuye en un 4.5 % a las 24 h, 22.7 % a las 96 h, 36.4% a los 10 días y 40.9% a los 20 días. (barra A a E= 300 μm, F= 400 μm).
Diez días postisquemia, en el hemisferio contralateral a la lesión, se observaron las láminas corticales sin cambios en la inmunoreactividad al compararlas con el control (Fig. 2 G y H). A los 20 días postisquemia el hallazgo es indiferenciable de los encontrados en los especímenes de 10 días que consiste en una inmunoreactividad al NeuN similar a los controles; diferente de lo observado en las muestras de 24 y 96 horas.
Una observación interesante consistió en que en el 60% de las ratas Winstar se presentaron alteraciones en la morfología de la lámina VI (Fig. 3), cambios que se detectaron de forma mas evidente a las 96 h postisquemia (Fig. 3 C) y que se continuaron observando hasta los 10 días (Fig. 3 D) y 20 días (Fig. 3 E), se pudo apreciar en ésta lámina una alteración de la citoarquitectura del tejido y una disminución y pérdida de la inmunoreactividad neuronal progresiva: con relación a la cuantificación de neuronas en los controles que definimos como proporción del 100%, el número de neuronas inmunoreactivas disminuyó al 95.5% y 77.3% a las 24 y 96 h respectivamente, a los 10 y 20 días la cantidad de neuronas inmunoreactivas corresponde al 63.6% y 59.1% respectivamente. En vista panorámica a 10x de aumento, la pérdida se observó en parches en algunos sectores de la lámina VI (Fig. 3 F).
Discusión
El análisis de los cambios en las neuronas del hemisferio contralateral a una lesión isquémica focal ha sido poco evaluado mediante el uso de marcadores específicos para la población neuronal 43-45. Nagasawa y Kogure 46 no encontraron anormalidades histológicas en el hemisferio contralateral después de una isquemia cerebral focal en un modelo experimental igual al nuestro. Utilizaron la tinción de Nissl y de hematoxilina que no es específica para la población neuronal 42. Por otro lado, se han encontrado diferencias en la susceptibilidad al infarto cerebral entre las diferentes cepas de ratas 47, lo que podría tener relación con la heterogeneidad de los resultados de diferentes investigaciones, sobre todo en lo referente a cambios exofocales contralaterales.
En este estudio se tuvo en cuenta la intensidad de la inmunoreactividad como medida fisiopatológica de daño cerebral isquémico como se ha señalado en otros trabajos 37,48. Los cambios más significativos se encuentraronn en áreas dorsolaterales en ambos hemisferios, regiones que sufren en este modelo mayor pérdida neuronal ipsilateral y cambios en la inmunoreactividad de las neuronas en corteza contralateral. En la región frontoparietal ipsilateral a la lesión, se observaron diversos grados de daño neuronal que van desde necrosis evidente a las 24 h, hasta pérdida total del tejido con formación de zonas quísticas, como se reporta en otros estudios 4,9,49.
Con relación a los cambios exofocales en corteza contralateral a la lesión, nuestro estudio tiene tres aspectos a resaltar. En primer lugar, a las 24 h después de la isquemia se evidenció disminución de la inmunoreactividad al NeuN en las láminas supragranulares de la corteza frontoparietal contralateral a la lesión, a las 96 h, los cambios son evidentes tanto en las láminas supragranulares como en las infragranulares. Llama la atención que algunas neuronas, a las 96 h, presentaron una inmunoreactividad que dan cuenta de una aparente fragmentación de los núcleos, hasta ahora no descritos con NeuN (Fig. 2 F). Es importante resaltar que en estudios previos han establecido una colocalización de neuronas con disminución de la inmunoreactividad al NeuN y prueba de túnel positiva 37.
En segundo lugar, 10 y 20 días después de la isquemia, la inmunoreactividad al NeuN previamente atenuada se restableció en las zonas supragranulares e infragranulares, pero se evidenció una alteración de apariencia residual en la lámina VI (Fig. 3) dado que permanece desde las 96 h hasta los 20 dias posterior a la lesión isquémica. Previamente se han descrito procesos de plasticidad cortical relacionados con conexiones tálamo-corticales dado que la respuesta de la corteza podría tener relación con las aferencias que provienen del tálamo a la lámina VI 20,50. Se plantea entonces que aparte de las señales de deaferentación del foco isquémico a través del cuerpo calloso, las aferencias talámicas tendrían un efecto en la reorganización cortical17, 19.
En tercer lugar, se ha señalado que los mecanismos de los cambios exofocales transcallosos involucran las conexiones comisurales y la modulación de las sinapsis 4,15,51. Se ha observado hiperexcitabilidad en el hemisferio contralateral y cambios en el metabolismo que podrían tener relación con estos cambios exofocales transcallosos 52. La disminución en la actividad de neuronas gabaérgicas del hemisferio lesionado y el incremento de la actividad glutamatérgica ocasionado por la isquemia se puede traducir en una sobreexcitación en el hemisferio no lesionado 26. De esta manera podría explicarse la reorganización de funciones motoras después de un ACV 51,52. Se ha descrito que en ratas sometidas a isquemia focal el córtex motor en el hemisferio no isquémico sufre un proceso de reducción de las espinas dendríticas, seguido de arborización en las láminas infragranulares. En el pico del crecimiento dendrítico, 18 días después de la isquemia, la densidad sináptica en las láminas infragranulares y la densidad de espinas es normal 16.
Este es un estudio descriptivo de la inmunoreactividad al NeuN en corteza contralateral a la lesión. Si bien nos permite observar una secuencia de eventos relacionados con la intensidad de la reacción inmunohistoquímica y apreciaciones morfológicas, el resultado debe ampliarse en estudios posteriores en los cuales se puedan evaluar las densidades ópticas y la cuantificación de neuronas. Faltan otros estudios además, que permitan ampliar el papel del anticuerpo NeuN como marcador de estrés celular y posible marcador de muerte celular.
Conclusión
Este estudio muestra de manera descriptiva los cambios en corteza cerebral contralaterales a la lesión después de una isquemia focal transitoria en el territorio de la arteria cerebral media en un modelo experimental en ratas. Se toma la inmunoreactividad al NeuN como el principal medio de análisis. Hasta el momento no se conocen estudios de diásquisis transcallosa con observación de dicho marcador. Se presentó específicamente variación en la intensidad de la inmunoreactividad al NeuN en función del tiempo desde las 24 h hasta los 20 días, observándose una respuesta subcrónica en zonas supragranulares y después infragranulares, es significativa la observación de cambios en la lámina VI que perduran desde las 96 h hasta los 20 días. En términos funcionales, la importancia de los cambios en la inmunoreactividad neuronal al NeuN podría estar asociado con procesos de reorganización celular adaptativa contralateral después del evento isquémico en la que participarían proyecciones cortico-corticales y cortico-talámicas, así como circuitos locales.