Introducción
La demencia es un síndrome caracterizado por la pérdida o deterioro de la memoria y otras funciones cognitivas como el habla, el lenguaje, el razonamiento, el juicio y el pensamiento. La alteración de estas funciones interfiere con la realización de actividades cotidianas. Se estima que en 2015 más de 46,8 millones de personas en todo el mundo tenían demencia y se espera que este número se duplique cada 20 años hasta alcanzar 131,5 millones en 2050 1. Estas cifras son probablemente subestimadas porque no incluyen a las personas en las primeras etapas de la enfermedad y los casos que son mal diagnosticados. Por estas razones, es probable que la demencia se convierta en uno de los problemas de salud más importantes del mundo.
La enfermedad de Alzheimer (EA) es la forma más frecuente de demencia de inicio en el adulto, caracterizada por deterioro progresivo de la cognición y la memoria, y representa entre el 60% y el 80% de los casos 2. El mayor número de individuos afectados se puede encontrar en regiones como Estados Unidos, Europa Occidental y China, y también en regiones en desarrollo como el Pacífico occidental y América Latina 3. Hay muchas otras causas de demencia incluyendo la enfermedad cerebrovascular, la demencia con cuerpos de Lewy (DCL), demencia mixta (EA y demencia vascular, EA y DCL, y la combinación de los tres), degeneración del lóbulo frontotemporal y enfermedad de Parkinson entre otras 2. Algunos aspectos de estas enfermedades se superponen entre sí haciendo difícil identificar la causa exacta, por lo que el diagnóstico preciso es una tarea compleja. Para diagnosticar adecuadamente la EA, deben descartarse otras formas de demencia. Esto incluye los trastornos metabólicos, endocrinos y nutricionales (Por ejemplo, enfermedad tiroidea, deficiencia de vitamina B12 e intoxicación por metales pesados); infecciones crónicas, tumores cerebrales, hematoma subdural, depresión y demencia inducida por medicamentos 4. La enfermedad tiene un progreso promedio de 7 a 10 años y aunque la duración es diferente en cada persona con EA, los síntomas parecen desarrollarse en las mismas etapas. Se ha planteado la hipótesis que los cambios en el cerebro comienzan 10 a 20 años antes de que aparezca cualquier manifestación clínica. Se ha establecido que la EA comienza con la muerte neuronal en la corteza entorrinal, una región conectada con el hipocampo que desempeña un papel importante en el aprendizaje y está involucrada en la transformación de las memorias a corto plazo en memorias a largo plazo. La atrofia de estas áreas del cerebro explica los síntomas de olvido observados en las primeras etapas de la enfermedad, sin embargo, otras alteraciones cognitivas, tales como cambios en la atención y la capacidad de resolver problemas están también presentes. La progresión de la demencia a un estadio leve dura de 2 a 5 años y se evidencia por pérdida de memoria, disfunción del lenguaje, dificultad visuoespacial, pérdida del conocimiento y cambios en la personalidad, entre otros. En este punto, la persona y la familia toman conciencia de la enfermedad y es cuando se efectúa el diagnóstico clínico. En la etapa moderada el daño se ha extendido a las regiones de la corteza cerebral que controlan el lenguaje, el razonamiento, el procesamiento sensorial y el pensamiento consciente. Los síntomas de la enfermedad se vuelven más pronunciados y la persona tiene problemas de conducta, por lo que se necesita más supervisión. En la EA, la interrupción de la comunicación neuronal puede causar alucinaciones, delirios, paranoia, estallidos de ira y también disminución de la capacidad para llevar a cabo tareas rutinarias (Por ejemplo, bañarse, vestirse, leer, escribir y trabajar con números). Esta es la etapa moderada de la EA y suele durar de 2 a 4 años. El estadio severo de la EA se caracteriza por una atrofia generalizada de la corteza cerebral y la ampliación de los ventrículos. La persona se vuelve completamente dependiente de los cuidadores porque es incapaz de reconocer a la familia y amigos. El individuo pierde la capacidad de tragar, controlar la función de la vejiga o el intestino, caminar y dormir.
Las causas de la EA siguen siendo desconocidas, pero la comunidad científica está de acuerdo en que múltiples factores están involucrados en la progresión de la enfermedad y que una sola causa es improbable. Se ha demostrado que varios factores de riesgo están relacionados con el desarrollo de la EA. La edad avanzada es el mayor factor de riesgo para la EA y la mayoría de los pacientes tienen 65 años o más 5. Otros factores de riesgo incluyen antecedentes familiares, ser un portador de alelo de la Apolipoproteína E-ε4 (APOE-ε4), tener deterioro cognitivo leve (MCI), factores de riesgo de enfermedad cardiovascular (colesterol alto, diabetes tipo 2, hipertensión arterial, tabaquismo, obesidad, etc.), y lesión cerebral traumática 6-10. Además de los factores antes mencionados, existen pruebas de que los factores de riesgo ambientales, como la calidad del aire, los metales pesados tóxicos y las exposiciones ocupacionales, también pueden contribuir al desarrollo de la EA 11.
La identificación de los factores de riesgo es un paso clave en el diagnóstico precoz de la EA. En la última década se han desarrollado varias herramientas para controlar el inicio y la progresión de la enfermedad. Cada vez hay más biomarcadores (biomoléculas específicas presentes en sangre o líquido cefalorraquídeo o técnicas de imagen) que permiten identificar los cambios celulares y cerebrales años antes de que comiencen los primeros síntomas clínicos de la demencia. Los biomarcadores actuales de la enfermedad se centran en medir los niveles de Aβ1-40, Aβ1-42 y la proteína Tau en el líquido cefalorraquídeo. Los estudios de imagen (MRI o PET) suelen complementar el análisis de biomarcadores de fluidos. La introducción del Compuesto Pittsburgh B (PiB) radiomarcado, que se une a las placas Aβ en el cerebro, ha permitido rastrear el proceso de agregación usando PET 12. Recientemente, se ha desarrollado la PET para la proteína Tau, este promete ser un buen biomarcador y puede ser útil para estimar la etapa de la enfermedad en la que se encuentra el paciente 13,14.
Hasta el momento no existe cura para la enfermedad, los tratamientos disponibles son sólo sintomáticos y la eficacia disminuye a medida que progresa la neurodegeneración. La inmunoterapia contra Aβ es uno de los enfoques más prometedores para modificar el curso de la EA, por lo que esta revisión discutirá los principales aspectos de la inmunización activa y pasiva describiendo los prototipos de fármacos evaluados en diferentes ensayos clínicos.
Enfermedad de Alzheimer: Patogénesis e Inmunoterapia
Neuropatogénesis de la Enfermedad de Alzheimer
Los signos patológicos más importantes de la EA son las placas seniles y los ovillos neurofibrilares (NFT). Los primeros son agregados extracelulares de péptidos Aβ y los últimos son agregados intracelulares de la proteína Tau hiperfosforilada, una proteína asociada a microtúbulos 15. Mientras que en la hipótesis de la cascada amiloide, la evidencia genética, patológica y bioquímica implica la agregación de Aβ como un desencadenante temprano crítico en la cadena de eventos que conducen a la tauopatía, la disfunción neuronal y la demencia 16, el grado de deposición de Tau se correlaciona con el deterioro cognitivo en la EA 17,18 cuestionando el papel de la deposición de Aβ como el desencadenante de la patogénesis de Tau. Inicialmente, la hipótesis amiloide declaró que la disfunción neuronal y la muerte se producían por los efectos tóxicos de la carga total de Aβ. Recientemente se ha sugerido que no sólo la eliminación de Aβ, sino también su producción pueden estar alteradas en pacientes con EA. Además, nuevos estudios indican que no sólo los péptidos Aβ (Aβ1-40 y Aβ1-42) contribuyen a la disfunción neuronal, sino que las formas oligoméricas de la proteína (pequeños agregados de dos a 12 péptidos) son en realidad más deletéreas a las funciones cerebrales que los agregados Aβ tales como placas seniles 19,20. Los péptidos Aβ también pueden crecer en fibrillas, que se disponen en láminas β para formar las fibras insolubles de las placas amiloides 21.
Los análisis post-mortem de cerebros humanos revelan una progresión característica de las placas de Aβ y un patrón regular de aparición de NFT. La progresión de la apariencia de las placas Aβ se correlaciona funcionalmente y anatómicamente con las regiones cerebrales afectadas 22,23. La agregación de Aβ afecta primero las capas II-V del isocortex, seguidas por la corteza entorrinal, la formación hipocampal, la amígdala, la corteza insular y cingulada; Se extiende entonces a los núcleos subcorticales, incluyendo el estriado, los núcleos colinérgicos del cerebro anterior basal, el tálamo, el hipotálamo y la sustancia blanca. Mientras que los NFT surgen primero en el locus coeruleus, corteza entorrinal y áreas límbicas del cerebro, tales como el subículo de la formación hipocampal, la amígdala, el tálamo y el claustro, y luego se extienden a regiones neocorticales interconectadas 18,24. La incidencia de placas y ovillos se correlaciona positivamente con la EA, pero hasta ahora no existe relación anatómica entre las lesiones.
Mecanismo molecular del procesamiento de la proteína precursora amiloide (APP)
La vía proteolítica implicada en el procesamiento de APP ha sido bien caracterizada usando varios modelos in vitro e in vivo25,26. La APP se produce en grandes cantidades en las neuronas y se metaboliza muy rápidamente 27. Después de su paso por el retículo endoplásmico (ER) y Golgi, la APP es transportada a los terminales axónicos y sinápticos. El procesamiento de la APP tiene lugar en la red trans-Golgi (TGN) y desde allí se puede transportar a la superficie celular o a compartimentos endosómicos. Ambos pasos están mediados por vesículas asociadas a clatrina. Una vez en la superficie celular, la APP puede ser proteolizada por α-secretasas y el complejo γ-secretasa en un proceso que no genera Aβ y que se conoce como la vía No amiloidogénica (Fig. 1). La otra posibilidad es que la APP puede reinternalizarse en vesículas recubiertas de clatrina en compartimentos endosómicos que contienen β-secretasas y el complejo γ-secretasa. El resultado de la interacción con estas enzimas es la producción de Aβ, que se libera entonces al espacio extracelular o se degrada en los lisosomas. Este proceso se conoce como la vía Amiloidogénica (Fig. 1) 28-30. La escisión de α-secretasa está mediada por miembros de la familia de proteínas del dominio desintegrina y metaloproteinasa (ADAM), siendo ADAM-9, -10, -17 y -19 los candidatos más probables para llevar a cabo esta función 31,32. El sitio de corte de α-secretasa se encuentra dentro de la secuencia Aβ y, por lo tanto, evita la formación de este 33. La actividad enzimática de α-secretasa genera dos fragmentos. El fragmento N-terminal se denomina APP alfa secretado (sAPPα) y el fragmento C-terminal (CTF) se denomina CTF83 debido a la cantidad de residuos de aminoácidos de este péptido (Fig. 1). La correspondiente escisión de CTF83 por el complejo γ-secretasa genera un péptido pequeño conocido como p334. La enzima de escisión de la APP de sitio beta 1 (BACE1) es la β-secretasa más importante en el cerebro y es responsable de la producción de los fragmentos sAPPβ y CTF99 (Fig. 1). El procesamiento subsiguiente de CTF99 por el complejo γ-secretasa conduce a la formación de Aβ y el dominio intracelular de APP amino-terminal (AICD) (Fig. 1) 34,35. Un grupo de proteínas constituye el complejo γ-secretasa. Para este complejo se requieren cuatro proteínas: PS1 o PS2, nicastrina, potenciador de presenilina 2 (PEN-2) y faringe anterior defectuosa-1 (APH-1). La γ-secretasa escinde APP en su región transmembrana para crear Aβ1-40/Aβ1-42 (Fig. 1) o p3 y AICD59/57, un segundo corte en el sitio de escisión ε produce el fragmento AICD5036,37.
Figura 1
Procesamiento de APP y epítopes de mAb en Aβ. En la vía No amiloidogénica, la APP se escinde primero por α-secretasa (ADAM-10) produciendo dos fragmentos, sAPPα y C83, la última es cortada por el complejo γ-secretasa generando los péptidos p3 y AICD. La vía Amiloidogénica implica el corte de APP por β-secretasa (BACE1) que produce los fragmentos sAPPβ y C99; C99 se procesa a continuación por el complejo γ-secretasa produciendo los péptidos Aβ y AICD. La Figura muestra la región del epítope dentro de la secuencia de Aβ reconocida por mAb derivados de secuencia.
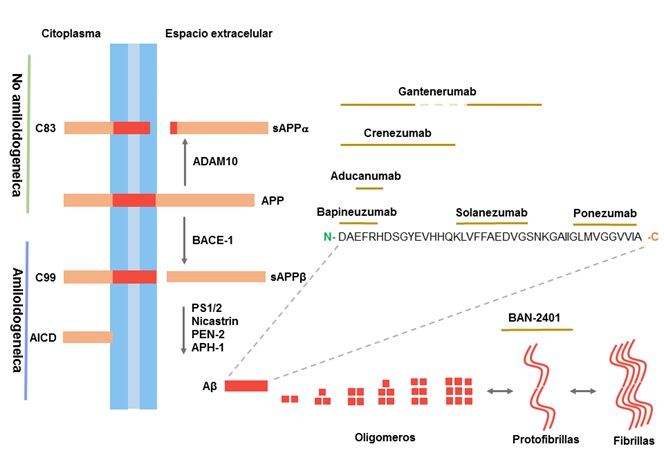
Péptidos Aβ y agregados
El péptido Aβ puede considerarse el principal producto del procesamiento proteolítico de APP. El péptido se encuentra tanto en el cerebro humano sano como en el cerebro humano afectado en concentraciones nanomolares o incluso más bajas. Hay varias isoformas de Aβ que se diferencian por el número de residuos de aminoácidos en la región C-terminal del péptido. La isoforma Aβ1-40 tiene 40 residuos y es la especie de Aβ más abundante en el cerebro de pacientes con EA 38. Aβ1-42 también está relacionado con la enfermedad porque es menos soluble que Aβ1-40 y forma agregados más rápido 39. Los péptidos Aβ tienen un tipo específico de disposición de hoja β que favorece la polimerización y la agregación, dando lugar a la formación de especies oligoméricas que se difunden a través de los fluidos intersticiales. Los monómeros Aβ tienden a agregarse y polimerizarse, formando oligómeros, protofibrillas y fibrillas (Fig. 1). Estudios han demostrado que estos ensambles surgen de Aβ de bajo peso molecular (monómeros o dímeros) 40,41. Sin embargo, hay controversia acerca de la toxicidad de las diferentes formas de Aβ. Los resultados indican que los conjuntos oligoméricos solubles pueden inhibir la actividad electrofisiológica que puede ser importante para la formación y el almacenamiento de la memoria, por lo que este paso parece crítico para el desarrollo de la EA 42. Los oligómeros también se unen a las subunidades del receptor N-metil-D-aspartato (NMDA) inhibiendo la plasticidad sináptica y alterando la homeostasis del calcio 43,44, lo cual causa la muerte neuronal. Por lo tanto, elegir como blanco terapéutico las formas de Aβ puede retrasar el proceso de agregación y mitigar la disfunción cognitiva que es el síntoma primario de la enfermedad.
Enfermedad de Alzheimer Esporádica y Familiar
Las dos variantes básicas de la EA son la esporádica (EAE) y la familiar (EAF). La EAE esporádica se caracteriza por la ausencia de patrón de herencia y de acuerdo con la edad de inicio se puede clasificar como de inicio precoz (antes de los 60 años de edad) o de inicio tardío (después de los 60 años de edad). La EAF se caracteriza por una heredabilidad autosómica dominante; representa probablemente menos del 1% de los casos de EA, y tiende a desarrollarse antes de los 60 años de edad (inicio temprano) 45,46. En la variante familiar la causa de la enfermedad es una mutación genética en los genes que codifican la proteína precursora amiloide (APP), presenilina-1 (PS1) o presenilina-2 (PS2) 47. En la actualidad, más de 32 diferentes mutaciones sin sentido se han encontrado en APP. Las mutaciones en APP representan del 10% al 15% de los casos de EAF y la mayoría tienen una edad de inicio de 45 años. Un número importante de las mutaciones se producen en el sitio de escisión de secretasa o en el dominio transmembrana. Ejemplos de ello son las mutaciones "Sueca" (K670N>M671L) y "Londres" (V717I) las cuales están entre las mutaciones más estudiadas de la APP que conducen al aumento de la producción de Aβ y al desarrollo de la EA 46. Con respecto a PS1, se han identificado más de 180 mutaciones responsables de alrededor del 80% de los casos de EAF. Estas mutaciones causan las formas más severas de EA; tienen una penetrancia completa y un inicio temprano de ~ 45 años 47,48. Las mutaciones en PS1 parecen aumentar la relación de Aβ1-42 a Aβ1-40 como resultado de un aumento de Aβ1-42 y disminución de la producción de Aβ1-4049. Hasta el momento se han identificado más de 390 familias portadoras de mutaciones PS1. Sin embargo, el grupo más grande del mundo de individuos que llevan una mutación sin sentido en PS1 consiste en alrededor 6.000 miembros de la familia colombiana que lleva la mutación E280A 50,51. En contraste con la mutación en el gen PS1, las mutaciones sin sentido en PS2 rara vez causan EAF de inicio temprano. La edad de inicio entre los miembros afectados de una familia tiene alta variabilidad 52. Actualmente se han identificado mutaciones en seis familias 53, una de ellas da como resultado la sustitución de una valina por una metionina en el residuo 393 (V393M) localizado dentro del séptimo dominio transmembrana 54.
La comprensión de la patogénesis de la EA ha sido muy influenciada por las cohortes de EAF. Sin embargo, la etiología de la EAE es heterogénea y muchos aspectos todavía son desconocidos. La falta de claridad sobre las bases moleculares de la enfermedad y la interacción de múltiples factores (Por ejemplo, genética, epigenética y medio ambiente) explican la complejidad de esta variante de la EA. La falta de herramientas precisas para el diagnóstico precoz, la incertidumbre para evaluar las etapas de la enfermedad y la rigidez del proceso clásico de descubrimiento de fármacos puede haber contribuido al fracaso de las estrategias terapéuticas empleadas durante las dos últimas décadas. Además, los compuestos desarrollados hasta ahora se centran en vías únicas, la mayoría de ellas dirigidas a la cascada amiloide. La evidencia emergente demuestra que otros cambios moleculares (Por ejemplo, la expresión de genes, el metabolismo lipídico, la actividad de Erk y otras quinasas) ocurren en paralelo con la agregación de Aβ y Tau incluso años antes de la aparición de los primeros síntomas 55-57. La comprensión de estos fenómenos debe ampliarse para tener éxito en la búsqueda de nuevas terapias contra la EA.
Tratamiento farmacológico estándar contra la EA
Durante las últimas décadas se han hecho muchos esfuerzos para encontrar el tratamiento definitivo para la EA. Las estrategias varían desde alternativas paliativas a estrategias más sofisticadas de modificación de la enfermedad, como la terapia génica y la inmunoterapia. Los tratamientos paliativos incluyen inhibidores de la enzima acetilcolinesterasa, que disminuyen la descomposición del neurotransmisor acetilcolina. Dentro de este grupo la rivastigmina, la galantamina y el donepezil están actualmente aprobados para su uso en la EA leve a moderada (Tabla 1) 58,59. Estos medicamentos parecen mejorar temporalmente la cognición, los síntomas conductuales y las tareas de rutina 60. Sin embargo, los ensayos clínicos han mostrado que un tercio de los individuos tenían beneficios mensurables, un tercio empeoró los síntomas durante los primeros 6 meses y el 29 % interrumpió el tratamiento debido a los efectos adversos 60-62. Otro fármaco aprobado para el tratamiento paliativo de la EA es la memantina, un antagonista del receptor NMDA, que reduce la neurotoxicidad mediada por estos mejorando así la cognición, el comportamiento y las actividades de la vida diaria (Tabla 1) 63. Se utiliza para la EA moderada a severo, pero a pesar de los beneficios observados, las altas tasas de abandono y la falta de efecto en la progresión de la enfermedad han limitado los ensayos que evalúan este fármaco 58,59,64. La eficacia limitada de los fármacos mencionados anteriormente y la necesidad de agentes terapéuticos capaces de modificar el curso de la enfermedad impulsaron la exploración de otros campos de la biomedicina que revolucionan el enfoque farmacológico clásico adoptado hasta ahora.
| Medicamento | Compañia de origen | Acción | Estado del paciente con EA | Eficacia |
|---|---|---|---|---|
| Donezepil | Eisai | AChI | Leve a moderado | Mejora cog., beh., DL |
| Rivastigmina | Novartis | AChI | Leve a moderado | Mejora cog., beh., DL |
| Galantamina | Janssen | AChI | Leve a moderado | Mejora cog., beh., DL |
| Memantina | Allergan | Antagonista MDAR | Moderado a severo | Mejora cog. |
| AChI: Inhibidor de Acetilcolinesterasa, NMDAR: Receptor de N-Metil-D-aspartato Cog: Cognición, Beh: Comportamiento, DL: Actividades de la vida diaria Fuente: http://www.clinicaltrials.gov | ||||
Inmunoterapia Activa vs Inmunoterapia Pasiva
Basándose en la teoría amiloide que sitúa Aβ como el primer y principal factor patógeno de la EA, los intentos de disminuir la causa de la neurodegeneración por las especies de Aβ tratan de programar el sistema inmunológico del propio paciente para deshacerse de los péptidos Aβ impidiendo la formación de placas amiloides. A esto se le llama inmunoterapia Aβ activa y utiliza la proteína sintética completa, o un fragmento de esta para estimular la producción de anticuerpos por las células B. Los anticuerpos neutralizan los péptidos Aβ y el complejo se elimina del cerebro. En 2002, la primera vacuna activa anti-EA (AN1792) desarrollada por ELAN en Irlanda y Wyeth en EE.UU pasó por un ensayo clínico de fase IIa. La vacuna contenía el péptido Aβ1-42 de longitud completa y mostró algunos efectos beneficiosos incluyendo menos declinación cognitiva. Sin embargo, el ensayo se suspendió debido al desarrollo de meningoencefalitis en ~ 6% de los individuos tratados con la vacuna 65-67. Una de las explicaciones más plausibles del desarrollo de este proceso inflamatorio es que uno de los excipientes utilizados en la preparación produjo la exposición de la región C-terminal de Aβ1-42, que parece activar la respuesta T-ayudador tipo 2 67. Por esta razón, las nuevas vacunas no incluyen esta región del péptido. Actualmente, se están desarrollando varias vacunas; estas incluyen CAD106 diseñada por Novartis en Suiza, ACI-24 creada por AC Immune en Suiza y UB-311 hecho por United Neuroscience Ltd en Irlanda. CAD106 contiene el péptido Aβ1-6 acoplado con un portador con copias del revestimiento proteico del bacteriófago QB para la inducción de la respuesta inmune (Tabla 2). Los ensayos de fase I no mostraron casos clínicos de meningoencefalitis. Sin embargo, durante los ensayos de fase IIa, un paciente presentó hemorragia intracerebral, mientras que cuatro individuos presentaron anomalías en imágenes diagnósticas que estaban relacionadas con Aβ 68,69. El ACI-24 está hecho con el péptido Aβ1-15 tetra-palmitoilado que favorece el plegado de las hoja β (Tabla 2). Este diseño es capaz de inducir la producción de anticuerpos específicos de conformación y se formula como membranas de liposomas para provocar la respuesta inmune 70. Se han iniciado estudios de fase I/IIa y se está a la espera de la publicación de los resultados preliminares (https://clinicaltrials.gov; Identificador: NCT02738450). UB-311 consiste en el péptido Aβ1-14 en combinación con el epítopo de células T ayudador UBITh(r), que induce específicamente la activación de células Th-1 (Tabla 2). La vacuna se probará en un estudio de fase IIa que evaluará la seguridad y la inmunogenicidad en pacientes con EA leve (https://clinicaltrials.gov; Identificador: NCT02551809). Aunque la inmunoterapia activa ha demostrado algunos beneficios para los pacientes con EA y hay grandes expectativas con respecto a los ensayos clínicos en curso, la seguridad y la dificultad para tratar los efectos adversos todavía causan preocupación y constituyen el principal inconveniente de este enfoque terapéutico.
| Inmunoterapia Activa | ||||||
|---|---|---|---|---|---|---|
| Vacuna | Compañia de origen | Blanco | Adyuvante de Formulación | Fase de ensayo clínico | Estado del paciente con EA | Resultado |
| AN1792 | ELAN/Wyeth | Aβ1-42 | QS-21, polisorbato 80 | IIa-terminado | Leve a moderado | Sin mejoría |
| CAD106 | Novartis | Aβ1-6 | Copias revestimiento proteico Bacteriófago QB | III | Prodrómico | NR |
| ACI-24 | AC Immune | tetra-palmitoilado Aβ1-15 (Conformación β) | Liposomas | II | Adultos con síndrome de Down | NR |
| UB-311 | United Neuroscience Ltd | Aβ1-14 | CpG/Alum | II | Leve | NR |
| Inmunoterapia Pasiva | ||||||
| mAb | Compañia de origen | Antigeno or Epitope /IgG | Especies reconocidas | Fase de ensayo clínico | Estado del paciente con EA | Resultado |
| Crenezumab | AC Immune/Genentech | Piroglutamato- Aβ1-15 (A)/hIgG4 | Oligómeros, fibrillas y placas | II | Leve | Disminuye niveles de Aβ |
| Bapineuzumab | Janssen/Pfizer | NT Aβ1-5 (E)/hIgG1 | Monómeros, fibrillas y placas amiloides | III | Leve a moderado | Estabiliza niveles de Aβ |
| Ponezumab | Janssen/Pfizer | CT Aβ1-40 (E)/hIgG2a | A(1-40(monómeros, oligómeros y fibrillas | II | Leve a moderado | Disminuye niveles de Aβ |
| Solanezumab | Eli Lilly | Aβ16-24 (E)/hIgG1 | Monómeros(oligómeros y fibrillas | III | Leve | Disminuye niveles de Aβ |
| Gantenerumab | Roche | NT Aβ1-10 and region central Aβ18-27 (E)/humano IgG1 | Monómeros, oligómeros y fibrillas | III | Prodrómico a leve | Disminuye niveles de Aβ |
| Aducanumab | Biogen | NT Aβ3-6 (E)/humano IgG1 | Oligómeros y fibrillas | Ib | Prodrómico a leve | Disminuye niveles de Aβ |
| BAN-2401 | Biogen/Eisai/ BioArctic | Aβ1-42 AM protofibrillas (A)/hIgG1 | Protofibrillas | I | Leve | NR |
| A: Antigeno; E: Epitope; hIgG: Humanizado IgG; NT: Región N-terminal; CT: Región C-terminal; AM: Mutación del Ártico; NR: No reportado Fuente: http://www.clinicaltrials.gov | ||||||
La inmunoterapia pasiva resuelve los problemas de la inmunización activa mediante el uso de anticuerpos monoclonales (mAb), que actúan a través de tres mecanismos que tienen lugar una vez que el anticuerpo ha cruzado la barrera hematoencefálica 71,72. El primero está mediado por la interacción Aβ-mAb que disminuye la formación de agregados tóxicos. El segundo requiere la unión entre el dominio Fc del mAb y los receptores Fc-γ presentes en la microglia que conduce a la fagocitosis del complejo Aβ-mAb. El tercer mecanismo implica la activación del efecto de citotoxicidad dependiente del complemento por el complejo Aβ-mAb que produce la lisis de la célula diana. Existe un cuarto mecanismo de acción en el que el mAb interactúa con Aβ en la sangre periférica y crea un gradiente de concentración que causa el flujo de Aβ desde el cerebro. Algunos mAb contra Aβ que están siendo probandos en ensayos clínicos incluyen bapineuzumab, ponezumab, solanezumab, gantenerumab, aducanumab, crenezumab y BAN-2401. Es importante anotar que la mayoría de estos ensayos clínicos utilizan un diseño aleatorizado que implica limitaciones como la falta de heterogeneidad entre los participantes y la dificultad que posee la evaluación cognitiva entre individuos con diferente sexo, edad y nivel educativo entre otros. A pesar de estas falencias, esta metodología es el "gold standard" para los estudios de eficacia del tratamiento y puede ser complementada para superar algunas de las limitaciones mencionadas anteriormente 73.
Bapineuzumab fue el primer anticuerpo que se probó en ensayos clínicos después de la terminación del estudio clínico AN1792. Consiste en un mAb IgG1 humanizado que se une a la región N-terminal de Aβ (Tabla 2) 74. El análisis de ensayos clínicos de fase II en pacientes de EA leve a moderada reveló una modesta mejora relacionada con la estabilización de la cantidad de Aβ. Sin embargo, algunos individuos tratados con el anticuerpo sufrieron edema reversible que se considera una anomalía de imagen relacionada con amiloide (ARIA-E) 75,76. Este evento y la falta de claros beneficios durante la fase III llevaron a la finalización de los ensayos clínicos.
El Ponezumab es un mAb IgG2a humanizado contra el epítope C-terminal de Aβ, que tiene una unión mucho más fuerte a Aβ1-40 que a otros monómeros, oligómeros o fibrillas (Tabla 2). Disminuye la carga amiloide a través de la salida de Aβ desde el hipocampo inducido por la reducción del péptido en el plasma 77. Los resultados de los ensayos clínicos no evidenciaron una mejoría significativa en el deterioro cognitivo de los pacientes con EA leve a moderada 78,79 y actualmente se está probando para el tratamiento de la angiopatía amiloide cerebral (CAA) (https://clinicaltrials.gov; Identificador: NCT01821118).
Solanezumab es la versión humanizada del mAb m266 IgG1 que se une a la región central de Aβ y tiene más afinidad por los monómeros que por las especies solubles y tóxicas en pacientes con EA leve (Tabla 2) 80. En un primer momento, los resultados de los ensayos clínicos de fase III no demostraron mejoras significativas en los individuos tratados con el anticuerpo 81. Un análisis complementario de los datos reveló menos deterioro cognitivo y funcional en pacientes con EA 82. El anticuerpo ha sido bien tolerado, pero se han observado ARIA-E en 16 individuos incluidos en ensayos doble ciego (EXPEDITION y EXPEDITION 2) y en un ensayo abierto de extensión (EXPEDITION-EXT) 83. Además, la magnitud de los beneficios está en el mismo nivel de los inhibidores de la acetilcolinesterasa (https://clinicaltrials.gov; Identificador: NCT02760602 y NCT02008357). El 23 de noviembre de 2016, Eli Lilly anunció que abandonaría los ensayos clínicos sobre el fármaco debido a que los resultados del estudio EXPEDITION3 demostraron que el solanezumab no fue capaz de frenar el deterioro cognitivo en pacientes con EA en comparación con los que recibieron placebo. Una explicación plausible del fracaso es que el anticuerpo podría quedar atrapado en la sangre y no alcanzar concentraciones terapéuticas en el cerebro 84.
Gantenerumab fue el primer mAb completamente humano diseñado para unirse con afinidad subnanomolar a un epítope conformacional en las fibrillas Aβ. Abarca aminoácidos N-terminales y centrales de Aβ y se une a monómeros, oligómeros y fibrillas en individuos con EA prodrómica a leve (Tabla 2) 85. El anticuerpo reduce la carga amiloide y activa la microglia evitando la formación de placas 85. Durante los ensayos clínicos de fase I, el anticuerpo fue seguro y bien tolerado; Sin embargo, algunos pacientes tratados con dosis altas desarrollaron ARIA transitoria 86. Los estudios de fase II indicaron que no hubo eficacia en la cohorte evaluada, pero el análisis post-hoc mostró un ligero beneficio en pacientes con progresión rápida. En este momento están en curso ensayos clínicos de fase III, estos incluyen un estudio para evaluar el efecto del anticuerpo sobre la seguridad, farmacocinética, cognición y funcionamiento en individuos con EA prodrómica (https://clinicaltrials.gov; Identificador: NCT01224106). Un ensayo para probar la eficacia y la seguridad del gantenerumab en pacientes con EA leve (https://clinicaltrials.gov; Identificador: NCT02051608); y un estudio de fase II/III para determinar si el anticuerpo mejora el resultado cognitivo de los participantes con EA (https://clinicaltrials.gov; Identificador: NCT01760005).
El aducanumab es un mAb IgG1 humano desarrollado a partir de una librería de células B creada a partir de individuos sanos de edad avanzada 87. El anticuerpo interactúa con la región N-terminal de Aβ y se une a oligómeros y fibrillas de sujetos con EA prodrómica a leve (Tabla 2) 87,88. El ensayo clínico de fase Ib mostró una mejoría de la disminución cognitiva, pero causó ARIA en pacientes con dosis altas de tratamiento. Las limitaciones de este estudio incluyeron tamaños de muestra pequeños, el uso del diseño escalonado de la dosis secuencial y careció de potencia debido puntos finales clínicos exploratorios. El ensayo demostró ser seguro y eficaz en el aclaramiento amiloide, pero los efectos positivos de la cognición fueron menos claros 87. Con base en el análisis interno de datos y los resultados prometedores, se decidió iniciar dos estudios de fase III establecidos para evaluar la eficacia del aducanumab en la disminución del deterioro cognitivo y funcional en los participantes con EA temprana. Los ensayos se extenderán hasta el año 2022, en 150 centros en América del Norte, Europa, Australia y Asia (https://clinicaltrials.gov; Identificador: NCT02477800 y NCT02484547). La expectativa por los resultados de estos ensayos es grande porque pueden poner a prueba la hipótesis amiloide.
El crenezumab, también conocido como MABT, es un anticuerpo humanizado dirigido contra la región media de Aβ que usa un isotipo IgG4 para reducir el riesgo de sobre-activación microglial. Reconoce monómeros, oligómeros y fibrillas Aβ, aunque tiene menos afinidad por los primeros (Tabla 2) 70,89. En la actualidad, la "Alzheimer's Prevention Initiative" (API) está reclutando a 300 individuos colombianos, 200 que portan la mutación E280A en PS1 y 100 no portadores 90. El propósito del estudio es evaluar la seguridad y la eficacia del anticuerpo en una fase preclínica de la EA (https://clinicaltrials.gov; Identificador: NCT01998841).
BAN-2401 es un mAb humanizado dirigido contra la APP que lleva la mutación E22G en Aβ (mutación del Ártico) 91. El anticuerpo es capaz de reconocer una conformación específica en las protofibrillas Aβ (Tabla 2) 91. El ensayo clínico de fase I demostró que el anticuerpo era seguro y no se observaron eventos adversos graves 92. Un estudio de fase II está actualmente reclutando a los participantes para determinar la eficacia clínica de BAN-2401 en el deterioro cognitivo leve y EA leve (https://clinicaltrials.gov; Identificador: NCT01767311).
Conclusiones
La inmunoterapia trabaja junto con el sistema inmune humano para neutralizar el proceso de agregación de las especies de Aβ. Actualmente puede ser el mejor enfoque para modificar la neurodegeneración y el deterioro cognitivo presente en la EA. No obstante, más estudios son necesarios para encontrar vacunas con mayor especificidad que no induzcan la respuesta autoinmune. Respecto a la inmunización pasiva, es necesario mejorar la eficiencia de los mAb para cruzar la barrera hematoencefálica, así como la reactividad cruzada y las alteraciones inflamatorias observadas en algunos pacientes. También es conveniente contemplar el uso de compuestos no inmunogénicos tales como aptámeros de ADN o ARN, que son pequeños fragmentos de oligonucleótidos con fuerte afinidad por diversos objetivos que van desde moléculas pequeñas a células y pueden superar los problemas observados con la inmunización 93.
Agradecimientos:
B-O agradece a la Universidad Icesi por la financiación del proyecto interno No. CA041341. FL agradece al programa de Sostenibilidad del CODI Universidad de Antioquia
Referencias
Ali G-C, Guerchet M, Wu Y-T, Prince M, Prina M. Chapter 2: The global prevalence of dementia Prince M, Wimo A, Guerchet M, Ali G-C, Wu Y-T, Prina M. World Alzheimer Report 2015. The Global Impact of Dementia. An analysis of prevalence, incidence, cost and trends. Alzheimer's Disease International (ADI):; London: 2015. p. 10–29 | |
Alzheimer's Association. Special Report: The personal financial impact of Alzheimer's on families Alzheimer's Association. 2016 Alzheimer's Disease Facts and Figures. Chicago: Alzheimer's Association; 2016. p. 58–67. https://www.alz.org/documents_custom/2016-facts-and-figures.pdf | |
WHO. The Epidemiology and Impact of Dementia: Current state and future trends. 2015. WHO/MSD/MER/15.3. p. 4. http://www.who.int/mental_health/neurology/dementia/dementia_thematicbrief_epidemiology.pdf | |
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH. The diagnosis of dementia due to Alzheimer´s disease recommendations from the National Institute on Aging-Alzheimer´s Association workgroups on diagnostic guidelines for Alzheimer´s disease. Alzheimers Dement. 2011;7(3):263–269 | |
van der Flier WM.Pijnenburg YA.Fox NC.Scheltens P. Early-onset versus late-onset Alzheimer´s disease the case of the missing APOE ?4 allele. Lancet Neurol. 2011;10(3):280–288 | |
Lautenschlager NT, Cupples LA, Rao VS, Auerbach SA, Becker R, Burke J. Risk of dementia among relatives of Alzheimer´s disease patients in the MIRAGE study What is in store for the oldest old? Neurology. 1996;46(3):641–650 | |
Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer´s disease. J Clin Psychiatry. 2007;68(4):613–618 | |
Scarabino D, Broggio E, Gambina G, Maida C, Gaudio MR, Corbo RM. Apolipoprotein E genotypes and plasma levels in mild cognitive impairment conversion to Alzheimer´s disease A follow-up study. Am J Med Genet B Neuropsychiatr Genet. 2016;171(8):1131–1138 | |
Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kåreholt I, Winblad B. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62(10):1556–1560 | |
Li W, Risacher SL, McAllister TW, Saykin AJ. Traumatic brain injury and age at onset of cognitive impairment in older adults. J Neurol. 2016;263(7):1280–1285 | |
Killin LO, Starr JM, Shiue IJ, Russ TC. Environmental risk factors for dementia a systematic review. BMC Geriatr. 2016;16(1):175 | |
Villemagne VL, Pike KE, Chételat G, Ellis KA, Mulligan RS, Bourgeat P. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192 | |
Saint-Aubert L, Almkvist O, Chiotis K, Almeida R, Wall A, Nordberg A. Regional tau deposition measured by [(18)F]THK5317 positron emission tomography is associated to cognition via glucose metabolism in Alzheimer´s disease. Alzheimers Res Ther. 2016;8(1):38 | |
James OG, Doraiswamy PM, Borges-Neto S. PET Imaging of Tau Pathology in Alzheimer´s Disease and Tauopathies. Front Neurol. 2015;6:38 | |
Perl DP. Neuropathology of Alzheimer´s disease. Mt Sinai J Med. 2010;77(1):32–42 | |
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer´s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608 | |
Wilcock GK, Esiri MM. Plaques, tangles and dementia A quantitative study. J Neurol Sci. 1982;56(2-3):343–356 | |
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259 | |
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–6453 | |
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration lessons from the Alzheimer´s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112 | |
Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101(5):1172–1184 | |
Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800 | |
Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF. Molecular, structural, and functional characterization of Alzheimer´s disease evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25(34):7709–7717 | |
Braak H, Del Tredici K. The pathological process underlying Alzheimer´s disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171–181 | |
Selkoe DJ. Toward a comprehensive theory for Alzheimer´s disease Hypothesis: Alzheimer´s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25 | |
Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer´s β-amyloid precursor protein. J Neurochem. 2012;120 1:9–21 | |
Lee J, Retamal C, Cuitiño L, Caruano-Yzermans A, Shin JE, van Kerkhof P. Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J Biol Chem. 2008;283(17):11501–11508 | |
Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87(4):1561–1565 | |
Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160(1):113–123 | |
Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615–29619 | |
Fahrenholz F, Gilbert S, Kojro E, Lammich S, Postina R. Alpha-secretase activity of the disintegrin metalloprotease ADAM 10 Influences of domain structure. Ann N Y Acad Sci. 2000;920:215–222 | |
Asai M, Hattori C, Szabó B, Sasagawa N, Maruyama K, Tanuma S. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301(1):231–235 | |
Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248(4959):1122–1124 | |
Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer´s disease. Mol Brain. 2011;4:3 | |
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P. Beta-secretase cleavage of Alzheimer´s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741 | |
Selkoe DJ, Wolfe MS. Presenilin running with scissors in the membrane. Cell. 2007;131(2):215–221 | |
St George-Hyslop P, Fraser PE. Assembly of the presenilin -/ε-secretase complex. J Neurochem. 2012;120 1:84–88 | |
Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer´s disease. J Biol Chem. 1992;267(24):17082–17086 | |
Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairí M, Maglia G, Carulla N. Aβ42 assembles into specific β-barrel pore-forming oligomers in membrane-mimicking environments. Proc Natl Acad Sci U S A. 2016;113(39):10866–10871 | |
Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A. Amyloid beta-protein fibrillogenesis Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274(36):25945–25952 | |
Harper JD, Wong SS, Lieber CM, Lansbury PT. Assembly of A beta amyloid protofibrils an in vitro model for a possible early event in Alzheimer´s disease. Biochemistry. 1999;38(28):8972–8980 | |
Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M. Neurotoxicity of Alzheimer´s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29(19):3408–3420 | |
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31(18):6627–6638 | |
Yang TT, Hsu CT, Kuo YM. Cell-derived soluble oligomers of human amyloid-beta peptides disturb cellular homeostasis and induce apoptosis in primary hippocampal neurons. J Neural Transm (Vienna). 2009;116(12):1561–1569 | |
Bertram L, Tanzi RE. Alzheimer's disease: one disorder, too many genes? Hum Mol Genet. 2004;13 Spec No 1:R135–R141 | |
Goate A, Hardy J. Twenty years of Alzheimer´s disease-causing mutations. J Neurochem. 2012;120 1:3–8 | |
Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FL, Mitra RD. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer´s disease families. PLoS One. 2012;7(2):e31039 | |
Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer´s disease associated with mutations of the presenilin-1 gene. J Neurol. 2006;253(2):139–158 | |
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G. Mutant presenilins of Alzheimer´s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3(1):67–72 | |
Lopera F, Ardilla A, Martínez A, Madrigal L, Arango-Viana JC, Lemere CA. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277(10):793–799 | |
Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer´s disease a retrospective cohort study. Lancet Neurol. 2011;10(3):213–220 | |
Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA. Alzheimer´s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5(7):985–988 | |
Jayadev S, Leverenz JB, Steinbart E, Stahl J, Klunk W, Yu CE. Alzheimer´s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133(4):1143–1154 | |
Lindquist SG, Hasholt L, Bahl JM, Heegaard NH, Andersen BB, Nørremølle A. A novel presenilin 2 mutation (V393M) in early-onset dementia with profound language impairment. Eur J Neurol. 2008;15(10):1135–1139 | |
Vélez JI, Lopera F, Patel HR, Johar AS, Cai Y, Rivera D. Mutations modifying sporadic Alzheimer´s disease age of onset. Am J Med Genet B Neuropsychiatr Genet. 2016;171(8):1116–1130 | |
Mapstone M, Cheema AK, Fiandaca MS, Zhong X, Mhyre TR, MacArthur LH. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. 2014;20(4):415–418 | |
Lachén-Montes M, González-Morales A, de Morentin XM, Pérez-Valderrama E, Ausín K, Zelaya MV. An early dysregulation of FAK and MEK/ERK signaling pathways precedes the β-amyloid deposition in the olfactory bulb of APP/PS1 mouse model of Alzheimer´s disease. J Proteomics. 2016;148:149–158 | |
Sorbi S, Hort J, Erkinjuntti T, Fladby T, Gainotti G, Gurvit H. EFNS-ENS Guidelines on the diagnosis and management of disorders associated with dementia. Eur J Neurol. 2012;19(9):1159–1179 | |
Ihl R, Bunevicius R, Frölich L, Winblad B, Schneider LS, Dubois B. World Federation of Societies of Biological Psychiatry guidelines for the pharmacological treatment of dementias in primary care. Int J Psychiatry Clin Pract. 2015;19(1):2–7 | |
Raina P, Santaguida P, Ismaila A, Patterson C, Cowan D, Levine M. Effectiveness of cholinesterase inhibitors and memantine for treating dementia evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379–397 | |
Cummings JL, Isaacson RS, Schmitt FA, Velting DM. A practical algorithm for managing Alzheimer´s disease what, when, and why? Ann Clin Transl Neurol. 2015;2(3):307–323 | |
Birks J. Cholinesterase inhibitors for Alzheimer´s disease. Cochrane Database Syst Rev. 2006;(1):CD005593 | |
McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev. 2006;(2):CD003154 | |
Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M. Effect of vitamin E and memantine on functional decline in Alzheimer disease the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311(1):33–44 | |
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54 | |
Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64(1):94–101 | |
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562 | |
Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer´s disease randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11(7):597–604 | |
Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer´s disease. Alzheimers Res Ther. 2015;7(1):23 | |
Muhs A, Hickman DT, Pihlgren M, Chuard N, Giriens V, Meerschman C. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc Natl Acad Sci U S A. 2007;104(23):9810–9815 | |
Morrone CD, Liu M, Black SE, McLaurin J. Interaction between therapeutic interventions for Alzheimer´s disease and physiological Aβ clearance mechanisms. Front Aging Neurosci. 2015;7:64 | |
Lichtlen P, Mohajeri MH. Antibody-based approaches in Alzheimer´s research safety, pharmacokinetics, metabolism, and analytical tools. J Neurochem. 2008;104(4):859–874 | |
Booth CM, Tannock IF. Randomised controlled trials and population-based observational research partners in the evolution of medical evidence. Br J Cancer. 2014;110(3):551–555 | |
Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94(4):1550–1555 | |
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–2070 | |
Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer´s disease treated with bapineuzumab a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–372 | |
La Porte SL, Bollini SS, Lanz TA, Abdiche YN, Rusnak AS, Ho WH. Structural basis of C-terminal β-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer´s disease. J Mol Biol. 2012;421(4-5):525–536 | |
Landen JW, Zhao Q, Cohen S, Borrie M, Woodward M, Billing CB. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol. 2013;36(1):14–23 | |
Miyoshi I, Fujimoto Y, Yamada M, Abe S, Zhao Q, Cronenberger C. Safety and pharmacokinetics of PF-04360365 following a single-dose intravenous infusion in Japanese subjects with mild-to-moderate Alzheimer´s disease a multicenter, randomized, double-blind, placebo-controlled, dose-escalation study. Int J Clin Pharmacol Ther. 2013;51(12):911–923 | |
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer´s disease. Proc Natl Acad Sci U S A. 2001;98(15):8850–8855 | |
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer´s disease. N Engl J Med. 2014;370(4):311–321 | |
Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H. Phase 3 solanezumab trials Secondary outcomes in mild Alzheimer´s disease patients. Alzheimers Dement. 2016;12(2):110–120 | |
Carlson C, Siemers E, Hake A, Case M, Hayduk R, Suhy J. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer´s disease. Alzheimers Dement (Amst). 2016;2:75–85 | |
Abbott A, Dolgin E. Failed Alzheimer´s trial does not kill leading theory of disease. Nature. 2016;540(7631):15–16 | |
Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F. Gantenerumab a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers. Dis. 2012;28(1):49–69 | |
Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207 | |
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M. The antibody aducanumab reduces Aβ plaques in Alzheimer´s disease. Nature. 2016;537(7618):50–56 | |
Kastanenka KV, Bussiere T, Shakerdge N, Qian F, Weinreb PH, Rhodes K, et al.. Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J Neurosci. 2016;36(50):12549–12558 | |
Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ J. Neurosci. 2012;32(28):9677–9689 | |
Ayutyanont N, Langbaum JB, Hendrix SB, Chen K, Fleisher AS, Friesenhahn M. The Alzheimer´s prevention initiative composite cognitive test score sample size estimates for the evaluation of preclinical Alzheimer´s disease treatments in presenilin 1 E280A mutation carriers. J Clin Psychiatry. 2014;75(6):652–660 | |
Tucker S, Möller C, Tegerstedt K, Lord A, Laudon H, Sjödahl J. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43(2):575–588 | |
Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G. Safety and tolerability of BAN2401--a clinical study in Alzheimer´s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8(1):14 | |
Qu J, Yu S, Zheng Y, Yang H, Zhang J. Aptamer and its applications in neurodegenerative diseases. Cell Mol Life Sci. 2016; |