
Gen SRY y ausencia de tejido testicular en una mujer 47XYY con disgenesia gonadal
John Jairo Rojas, M.D.1, William Jubiz, M.D.2, Carolina Isaza, M.D., M.Sc.3
1. Residente de Ginecología y Obstetricia IV año, Facultad de Salud, Universidad Libre, Cali.
2. Profesor de Medicina Interna y Ginecología Endocrina, Facultad de Salud, Universidad Libre y Director Centro Endocrinología Metabolismo y Diabetes, Cali.
3. Profesora Titular, Departamento Morfología, Facultad de Salud, Universidad del Valle, Cali.
Recibido para publicación abril 20, 2004 Aprobado para publicación enero 28, 2005
RESUMEN
Este artículo revisa conceptos actuales sobre determinación y diferenciación sexual con base en el estudio genético de una niña de 13 años que consultó por talla baja y aumento de peso. El examen físico mostró Tanner I en mamas y en vello púbico, sin signos de androgenización. Mientras el nivel de la hormona de crecimiento (GH) era normal, las hormonas folículoestimulante (FSH) y luteinizante (LH) estaban aumentadas. Mediante laparoscopia y posterior estudio patológico se demostró la presencia de gonadas rudimentarias con ausencia de tejido testicular. Aunque el cariotipo obtenido fue 47XYY y el análisis molecular identificó la presencia del gen SRY, su funcionalidad es incierta, lo que hace necesaria su secuenciación, con la finalidad de determinar posibles mutaciones. En respuesta a la terapia con estrógenos y progesterona se desarrollaron tanto los caracteres sexuales secundarios como una menstruación normal. Aunque es posible que en la paciente haya una doble alteración genética donde concurran la mutación de novo de un gen y una no disyunción en la meiosis paterna, el caso descrito es ilustrativo de la importancia del estudio genético en la evaluación de la disgenesia gonadal.
Palabras clave:Disgenesia gonadal; Diferenciación sexual; Cariotipo 47XYY; Gen SRY; Hipogonadismo primario.
SUMMARY
This paper reviews current concepts about sex determination and differentiation in males and females. We also present a 13 year old girl, who consulted for short stature and weight gain. She was a Tanner I for breast and pubic hair. There were no signs of androgen excess. Lab showed a high FSH and LH with normal growth hormone. Laparoscopy revealed streak gonads without testicular tissue. Karyotype was 47XYY, with the SRY gene present. Functional studies of the gene to detect mutations were not performed. Patient responded to a combination of estradiol and medroxyprogesterone with normal menses and breast development. This patient is an example of the importance of performing genetic studies in the evaluation of gonadal dysgenesis cases. It is possible that a de novo mutation of the gene, rather than a paternal meiotic nondysjunction was present in our patient.
Key words:Gonadal dysgenesis; Sex differentiation; 47 XYY karyotype; SRY gene; Primary hypogonadism.
Los avances en la genética, la endocrinología experimental, la biología molecular y la bioquímica han permitido comprender las bases del desarrollo sexual. Este se inicia en el mismo momento de la fecundación, cuando se establece el sexo cromosómico del feto, según el cromosoma sexual (X o Y) aportado por el espermatozoide.
En la tercera semana de desarrollo se originan las células germinativas primordiales, a partir del endodermo del saco vitelino. Después de migrar durante la cuarta y quinta semanas a través del mesenterio dorsal, llegan a las gónadas primitivas, abultamientos localizados en las crestas urogenitales. El desarrollo de las crestas obedece a la acción de un grupo de genes como WT1, SF1, DAX1, Lim1, EMX2, LHX9, situados en cromosomas autosómicos1.
Las gónadas indiferenciadas primitivas se originan a partir del reborde epitelial celómico de la cresta urogenital y del mesodermo intermedio, ventral al mesonefros. Los cordones sexuales primarios crecen a partir del epitelio celómico y se dirigen en dirección dorsal, para penetrar en la gónada primitiva. La llegada de las células germinativas a la gónada indiferenciada durante la quinta semana de desarrollo favorece su desarrollo y diferenciación. Por tanto, en su ausencia, desaparece el efecto inductor sobre las gónadas y se produce agenesia gonadal.
Por otra parte, la determinación del sexo gonadal ocurre en las semanas 6 a 9 de desarrollo. La importancia del cromosoma Y en el desarrollo testicular fue establecida durante la década de 1950, mientras que la del antígeno H-Y, presente en el brazo largo del cromosoma Y, fue dilucidada una década más tarde.
Mediante el análisis de las alteraciones estructurales del cromosoma Y fue posible, en la década de 1980, identificar al gen responsable de la diferenciación testicular. Fue así como se postuló que correspondía al gen ZFY (Zinc Finger Chromosome Y), codificante de una proteína reguladora de transcripción y localizado en el brazo corto del cromosoma Y. Sin embargo, el locus del verdadero gen responsable de la diferenciación testicular, sólo se identificó a finales del siglo XX, después de estudios complejos basados en análisis genéticos moleculares de secuencias de traslocación y deleción, en pacientes 46XX y 46XY con inversión sexual2,3. El locus propuesto estaba en Yp11.3, inmediatamente proximal a la región pseudoautosómica del brazo corto del cromosoma Y. Al gen se le denominó SRY (Región Determinante del Sexo en el Cromosoma Y).
El factor de transcripción codificado por el gen SRY tiene un sólo exon de 850 kilobases. Su dominio funcional, denominado también grupo de alta movilidad (HMG), tiene 79 aminoácidos y se une al ADN para dar inicio a la transcripción del gen.
Desde el punto de vista evolutivo, la caja HMG del SRY es altamente conservada. Por fuera de la caja HMG existen ciertas regiones que podrían ser homologadas entre especies de animales con secuencias de SRY y seres humanos. Este hecho sugiere que dichas regiones no tienen significado funcional o bien, pueden ser muy específicas2.
Mediante la transcripción del gen SOX9 tanto en los pliegues gonadales vecinos del conducto de Müller como en los conductos metanéfricos, se ha logrado activar al gen SRY presente en las células del epitelio celómico, precursoras de las células de Sertoli. Por su parte,el desarrollo de las células de Leydig está en apariencia determinado por la expresión de WNT4.
La ausencia de SRY y la doble dosis de los genes del cromosoma X aseguran el desarrollo y la permanencia de los ovarios. El desarrollo ovárico es más tardío que el testicular. El epitelio celómico del embrión femenino, sometido a la actividad de los genes WNT4, DAX1 y FOXL2 da origen a las células de la granulosa. Esto hace no sólo que la gónada aumente su tamaño, sino también que las ovogonias sufran mitosis de forma acelerada. Entre las semanas 11 y 12, las ovogonias se diferencian en ovocitos primarios y se rodean de células granulosas, para formar así los folículos primordiales que se observan en el ovario aproximadamente durante la semana 14 y adquiere un máximo grado de desarrollo entre las semanas 20 y 25. En este momento, ya es posible reconocer claramente los rasgos morfológicos del ovario, lo mismo que algunos folículos primarios, formados a partir de los folículos primordiales.
Hacia la semana 12 de la gestación, tanto genitales internos como externos han concluido su proceso de diferenciación y forman en conjunto lo que se denomina �sexo genital�. En la diferenciación de los genitales internos toman parte, tanto los conductos mesonéfricos o de Wolff, como los paramesonéfricos o de Müller. Ambos tipos de conductos requieren de la estimulación de las hormonas gonadales para su diferenciación
Cuando hay desarrollo testicular, las células de Leidyg inician la producción de testosterona hacia la semana 9, bajo la influencia de la gonadotropina coriónica. Por su parte, las células de Sertoli comienzan a secretar la hormona antimülleriana. La secreción paracrina de testosterona actúa sobre los conductos de Wolff, de modo que estos dan origen a los conductillos eferentes, al epidídimo, al conducto deferente y a las vesículas seminales. La regresión de los conductos de Müller, provocada a su vez por la hormona antimülleriana, deja como remanentes al utrículo prostático y al apéndice del testículo4.
En la mujer, la ausencia de testosterona determina la regresión de los conductos mesonéfricos o de Wolff. En este caso, las estructuras remanentes son el paraóforo y el epoóforo. Debido a la ausencia de la hormona antimülleriana se desarrollan los conductos de Müller. Estos crecen en dirección caudal y se fusionan en la línea media para dar origen a las trompas de Falopio, al útero y a la parte superior de la vagina.
Los genitales externos se originan a partir de un esbozo común, de acuerdo con la disponibilidad de dihidrotestosterona. Cuando hay tejido testicular, la testosterona secretada es transformada en dihidrotestosterona, gracias a la acción de la enzima 5 a reductasa. Como resultado de su acción hay crecimiento del tubérculo genital y fusión de los pliegues del seno urogenital para dar origen al pene y a la uretra peneana. Los pliegues labioescoriales migran hacia la línea media donde se fusionan para formar el escroto. Debido a la ausencia de dihidrotestosterona en la mujer, los pliegues del seno urogenital quedan abiertos, de modo que se forman los labios menores. Los labios mayores a su vez derivan de los pliegues labioescrotales, mientras que el clítoris aparece a consecuencia del pobre crecimiento del tubérculo genital5,6.
Como complemento de esta revisión, se presentan la historia clínica, los hallazgos físicos, los resultados de las pruebas de laboratorio y la respuesta al tratamiento de una paciente con disgenesia gonadal.
DESCRIPCIÓN DEL CASO CLÍNICO
Paciente de 13 años, fruto de un primer embarazo sin complicación alguna. Talla al nacimiento de 51 cm. Detención del crecimiento a los 10 años de edad, según informe de la madre. A partir de ese momento, aumento progresivo de peso (en comparación con otros niños de la misma edad). Ausencia de desarrollo sexual y de menstruación.
En el Cuadro 1 se describen los hallazgos físicos y de laboratorio.
La edad ósea por carpograma fue 13 años. La ecografía pélvica informó las siguientes medidas del útero: longitudinal, 38 mm; anteroposterior 15 mm; transverso 22 mm. El ovario izquierdo midió 10 x 3 x 6mm. No se observó ovario derecho.
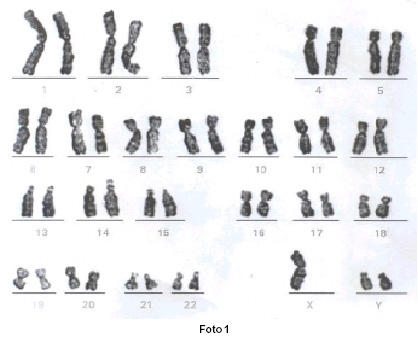
En la laparoscopia se observó útero de tamaño pequeño y forma normal, ovarios de apariencia rudimentaria, que fueron resecados quirúrgicamente. En el informe patológico se encontró tejido ovárico atrófico en ambas gónadas y ausencia de tejido testicular. El diagnóstico definitivo fue disgenesia gonadal 47XYY.
La paciente se trató con combinación de etinilestradiol 50 mg y levonorgestrel 250 mg, diariamente por 21 días, con una semana de descanso. Después del primer mes de tratamiento ocurrió la primera menstruación y a los tres meses se inició el desarrollo de los caracteres sexuales secundarios, tales como crecimiento de las mamas, piel fina y voz femenina (Foto 1).
DISCUSIÓN
La paciente consultó a los 13 años de edad, por retardo del crecimiento. Nunca había menstruado. Se descartó deficiencia de la hormona del crecimiento y se confirmó la presencia de un hipogonadismo primario (insuficiencia ovárica primaria). Con el hallazgo de un cariotipo 47XYY, se esperaba encontrar tejido testicular en la biopsia de la gónada. Sin embargo, en razón de la presencia del gen SRY, demostrada mediante análisis genético, la ausencia testicular indica una mutación en su estructura.
La literatura disponible informa 15 casos similares (45X/47XYY): ocho mujeres, tres varones y cuatro hermafroditas7. Este mosaicismo se puede explicar por medio de dos mecanismos posibles:
1. Deleción del gen SRY del cromosoma Y durante la recombinación meiótica con el cromosoma X
2. Mutaciones inactivantes del gen SRY. Se han informado más de 30 mutaciones inactivantes, la mayoría agrupadas en el ADN de la HMG y en su totalidad asociadas con XY reverso7.
El hipogonadismo hipergonadotrópico y la pubertad retrasada se pueden asociar con un cariotipo 46XX, así como con disgenesia gonadal 46XY. Mientras que las mujeres con síndrome de Turner tienen conductos müllerianos normales y gónadas rudimentarias, en la disgenesia gonadal pura 46XY se observan genitales ambigüos en combinación con pubertad retrasada. Además, en 15% de dichos individuos, se han encontrado mutaciones del gen SRY cuando se efectúa la determinación sexual de la región Y. SRY es una prueba determinante de la localización del gen en el brazo corto del cromosoma Y8.
REFERENCIAS
1. MacLaughlin D, Danahoe P. Mechanics of disease. Sex determination and differentiation. N Engl J Med 2004; 350: 367-378.
2. Teixeira J, Maheswaran S, Donahoe P. Müllerian inhibiting substance: an instructive developmental hormone with diagnostic and possible therapeutic applications. Endocr Rev 2001; 22: 657-674.
3. Berta P, Hawkins JR, Sinclair AH. Genetic evidence equating SRY and the testes-determinating factor. Nature 1990; 348: 448-450.
4. Haqq CM. Molecular basis of mammalian sexual determination: activation of müllerian inhibiting substance gene expression by SRY. Science 1994; 266:1494-1498.
5. Speroff L, Glass R, Kase N. Clinical gynecologic endocrinology and infertility. 6th ed. Philadelphia: Lippincott Williams & Williams; 1999. p. 107-113.
6. Page DC, Mosher R, Simpson EM, et al. The sex determining region of the human Y chromosome encodes a finger protein. Cell 1987; 51: 1091-1094.
7. Takagi A, Imai A, Tamaya T. A novel sex determining region on Y (SRY) nonsense mutation identified in a 45X/47XYY female. Fertil Steril 1999; 72: 167-169.
8. Gracia C. Molecular basis of puberal abnormalites. Obstet Gynecol Clin North Am 2003; 30: 261-267.