Descripción de un caso de síndrome de Cornelia de Lange. Aporte para un mejor diagnóstico pre- y post-natal
Martha Lucía Montes, M.D.1, Wilmar Saldarriaga, M.D.2, Carolina Isaza, M.D.3
1. Residente, Departamento de Patología, Escuela de Medicina, Facultad de Salud, Universidad del Valle, Cali, Colombia. e-mail: chiamontes@yahoo.com
2. Profesor Auxiliar, Departamentos de Morfología y Gineco-Obstetricia, Escuela de Medicina, Facultad de Salud, Universidad del Valle, Cali, Colombia. e-mail: wsaldarriaga0608@yahoo.com
3. Profesora Titular, Departamento de Morfología, Escuela de Medicina, Facultad de Salud, Universidad del Valle, Cali, Colombia. e-mail: carolinaisa@uniweb.net.co
Recibido para publicación marzo 9, 2005 Aceptado para publicación septiembre 8, 2006
RESUMEN
Introducción: El síndrome de Cornelia de Lange es poco frecuente, pero tiene múltiples malformaciones. Se caracteriza por hipertricosis en la cara, el labio superior delgado, micromelia, restricción del crecimiento intrauterino y retraso en el desarrollo postnatal. Este síndrome presenta expresividad variable y su etiología aún se desconoce. Casi todos los casos son esporádicos, y su diagnóstico es fundamentalmente clínico.
Caso clínico: Recién nacido de madre primigestante de 16 ańos, con embarazo de 35 semanas. En las ecografías obstétricas se encontró restricción del crecimiento intrauterino simétrico severo, oligohidramnios y compromiso en la circulación de las arterias umbilical y cerebral media. Por estos hallazgos se le hizo cesárea en el Hospital Universitario del Valle. El recién nacido falleció a los 20 minutos; 6 horas después se efectuó la autopsia.
Discusión: Los hallazgos en la autopsia fetal, los rayos X, el cariotipo y la historia clínica de la madre permitieron hacer el diagnóstico de síndrome Cornelia de Lange tipo I o forma clásica. Con base en la revisión de la literatura, se seleccionaron las características que sugieren la identificación en la ecografía obstétrica y el examen físico del recién nacido para hacer un mejor diagnóstico.
Palabras clave: Síndrome de Brachmann; Síndrome de Cornelia de Lange; Anomalías en extremidades superiores; Restricción de crecimiento intrauterino; Autopsia perinatal.
Description of a case of Cornelia de Lange syndrome. Contribution to better ante-natal and post-natal diagnoses
SUMMARY
Introduction: Cornelia of Lange syndrome is an unusual polimalformative disorder that shows facial hypertrichosis, thin upper lip, micromelic extremities, intra-uterine growth restriction and postnatal growth retardation. This syndrome shows wide phenotypical findings and its etiology remains unknown. Most of the cases are sporadic and the diagnosis is fundamentally a clinical one.
Clinical case: This is the case of a new born product of a G1 P0 A0, 16 year-old mother on her third trimester of pregnancy. Ultrasound showed symmetrical and severe intra-uterine growth restriction, oligohydramnios and altered circulation of the umbilical and cerebral arteries. Because of these findings, a cesarean section was performed at the Hospital of Universidad del Valle. The new born died after twenty minutes, and the autopsy was done 6 hours later.
Discussion: The findings of the autopsy, radiology, kariotype and mother’s clinical history indicated that this corresponded to a Cornelia of Lange syndrome type I or classic type. In accordance with literature review, features suggesting this entity in the obstetric sonography and in physical examination of the new born were selected since they can lead to an accurate diagnosis.
Keywords: Brachmann’s syndrome; Cornelia de Lange syndrome; Upper extremity abnormalities; Intrauterine growth restriction; Perinatal autopsy.
El primer caso de síndrome Brachmann-de Lange o Cornelia de Lange (SCDL), fue descrito por Brachmann en 19161 y posteriormente en 1933 Lange describió dos casos con similares características2, por lo cual este síndrome lleva sus nombres.
El síndrome es poco frecuente, con una prevalencia de 0.6 por 100,000 nacidos vivos (NV)3, y difiere según las poblaciones estudiadas. En Espańa la prevalencia es de 0.97 por 100,000 NV4 y en Estados Unidos es de 1 por 10,000 NV5.
El diagnóstico de este síndrome es fundamentalmente clínico; se basa en el reconocimiento de los rasgos fenotípicos faciales característicos, restricción del crecimiento intrauterino, anormalidades en las extremidades, retardo del desarrollo postnatal y retardo mental6.
El fenotipo facial se caracteriza por microbraquicefalia, implantación baja del cabello, pestańas largas, hipertricosis con sinofris, implantación baja de orejas, puente nasal bajo, fosas nasales antevertidas, filtro largo y boca en forma de carpa con labios finos, prognatismo maxilar y micrognatia.
Las anomalías de los miembros superiores pueden ser mayores o menores según el grado de discapacidad que produzcan, como micromelia, focomelia, implantación proximal de los pulgares, oligodactilia, agenesia o hipoplasia del cúbito, sindactilia y línea simiana. El llanto es típico y algunos pacientes pueden presentar convulsiones. También se han observado anomalías oculares, esqueléticas, cardíacas, digestivas, neurosensoriales, dermatológicas, genitales y endocrinas7,8. El SCDL se asocia, además, con alteraciones del comportamiento9,10.
La expresión de estas características es variable por lo cual Berney et al.9 propusieron clasificar los pacientes según la severidad de los hallazgos en tipo I o clásico y tipo II o leve.
En la forma clásica (tipo I) del SCDL, los pacientes presentan restricción de crecimiento intrauterino (RCIU), retardo psicomotor moderado a profundo y malformaciones mayores que producen discapacidades severas o la muerte. En el tipo II o forma leve, las características faciales y anormalidades esqueléticas menores son similares a las del tipo I; sin embargo, los problemas funcionales del síndrome pueden desarrollarse en el tiempo o expresarse parcialmente. Por lo general hay un retardo psicomotor leve a limítrofe, una restricción del crecimiento pre y postnatal menos severo y ausencia de malformaciones mayores11.
La etiología del síndrome Cornelia de Lange no se conoce. Casi todos los casos son esporádicos pero se han descrito algunos familiares con patrones de herencia autosómica dominante y autosómica recesiva12.
Los estudios citogenéticos son normales en la mayoría de los pacientes, aunque se han documentado múltiples casos con alteraciones en el cromosoma 3, algunos con duplicación especifica del área q21-qter y otros con deleciones del brazo largo. También se sabe de casos familiares que presentan una translocación equilibrada con puntos de quiebre en 3q26.3 y 17q23.1, por lo cual algunos autores proponen que el gen causante del SCDL se puede localizar en la región 3 q26.3. Hay informes de otras translocaciones equilibradas de novo, que se asocian con el SCDL como 5p13.1 y 13q12.1, y que comprometerían otros genes diferentes a los del cromosoma 313.
Recientemente Tonkin et al.14, en estudios de biología molecular en 4 casos esporádicos y 2 familiares del SCDL, informaron mutaciones en el gen NIPBL (Nipped-B gene like) localizado en la región 13.2 del brazo largo del cromosoma 5. Este gen codifica una proteína que promueve la expresión de otros genes pero se desconoce su función exacta para regular el desarrollo. Los autores nombraron esta proteína como delangina y es homóloga con la familia de las adherinas cromosomales con papeles diversos en la cohesión de cromátides hermanas, la condensación de los cromosomas y la reparación del ADN14.
Algunos autores han propuesto dentro de la clasificación considerar el SCDL tipo III para los casos donde haya relaciones con exposición a agentes teratógenos, deleción del ADN mitocondrial o anomalías cromosómicas15.
El objeto de este trabajo es agregar a la bibliografía existente un caso más de síndrome de Cornelia de Lange asociado con hernia diafragmática, y sugerir características fenotípicas que se deberían tener en cuenta para hacer el diagnóstico in útero y al nacimiento. Además, revisar los aspectos actuales de más interés en este síndrome.
DESCRIPCIÓN DEL CASO
Recién nacido de madre primigestante de 16 ańos, con embarazo de 35 semanas, a quien se le realizaron 5 ecografías obstétricas en las cuales se encontró RCIU simétrico severo, oligohidramnios y un aumento de la resistencia y del índice de pulsatilidad en las arterias umbilical y cerebral media y un perfil biofísico fetal de 4/8.
En el Hospital Universitario del Valle (HUV) por los hallazgos ecográficos, se programó cesárea, y se obtuvo un recién nacido de sexo femenino, quien falleció a los veinte minutos. Los padres autorizaron la autopsia neonatal que se hizo con los protocolos establecidos por la institución.
Autopsia neonatal. Estudios complementarios:
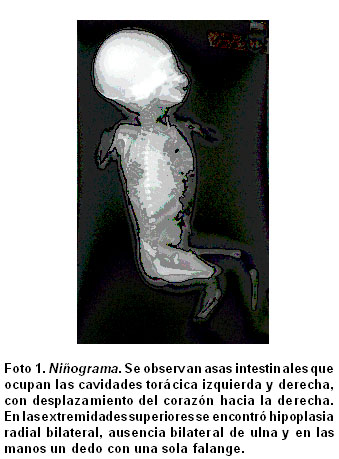
Radiografía AP y lateral neonatal. Se observaron asas intestinales que ocupaban las cavidades torácica izquierda y derecha, con desplazamiento del corazón hacia la derecha. En las extremidades superiores había hipoplasia radial bilateral, ausencia bilateral de ulna y en las manos un dedo con una sola falange (Foto 1).
Cariotipo: En las 20 metafases estudiadas con bandas G de rutina a una resolución de 800 bandas, se encontró un complemento cromosómico de 46, XX, sin anormalidades estructurales aparentes.
Hallazgos anatómico-patológicos. Recién nacido de sexo femenino malformado, sin esfacelos, con peso de 1,400 g; talla, 41 cm; perímetro cefálico, 28 cm; perímetro torácico, 26 cm; perímetro abdominal, 20 cm; todos por debajo del percentil 5 para la edad gestacional, lo que corresponde a una restricción severa y simétrica del crecimiento intrauterino.
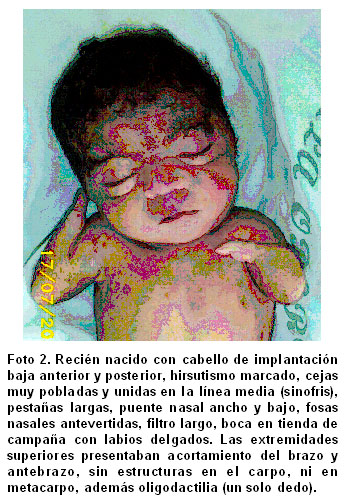
Facies dismórfica, cabello de implantación baja anterior y posterior, hirsutismo marcado, cejas muy pobladas y unidas en la línea media (sinofris), pestańas largas, puente nasal ancho y bajo, fosas nasales antevertidas, filtro largo, boca en tienda de campańa con labios delgados (Foto 2). Hendidura central a nivel de la porción dura del paladar secundario y micrognatia.
Las extremidades superiores presentaban acortamiento del brazo y antebrazo, sin movilidad en la articulación del codo, sin carpo ni metacarpo y oligodactilia (un solo dedo) (Foto 2).
Las extremidades inferiores no tenían alteraciones anatómicas aparentes.
En el examen interno se encontró hernia diafragmática izquierda a través de la cual protruían hacia ambos hemitórax 90% del intestino delgado, 50% del intestino grueso, el estómago, el bazo y el lóbulo izquierdo del hígado, los cuales desplazaban el corazón hacia la derecha y colapsaban totalmente el pulmón izquierdo y parcialmente el derecho. Los pulmones presentaron un número anormal de lóbulos. En la disección del corazón y los grandes vasos no se encontraron anormalidades.
En el abdomen no se evidenció hiperplasia suprarrenal ni anomalías del sistema urinario. Genitales externos femeninos, útero bicorne, con cérvix, tubas y ovarios normales.
DISCUSIÓN
Desde el principio de la consulta de dismorfología en el HUV en el ańo 1987, se ha hecho el diagnóstico a 9 pacientes con el síndrome de Cornelia de Lange (SCDL). En el ańo 2004 nacieron en el Hospital Universitario del Valle 2 nińos con las características del síndrome entre 7,000 nacimientos. En Colombia no hay datos de la prevalencia de este síndrome, aunque en la literatura se informa una prevalencia global de 0.6 por 100,000 NV para el SCDL3. Existen diferencias en las poblaciones estudiadas; en Espańa la prevalencia es de 0.97 por 100,000 NV4 mientras que Estados Unidos es de 1 por 10,000 NV5. Esta variación se puede deber a las dificultades para realizar el diagnóstico, pues éste se basa en los rasgos fenotípicos que son de expresión variable y no se cuenta con una prueba diagnóstica confirmatoria.
En el caso que se describe, los hallazgos frecuentemente asociados con SCDL se evidenciaron en el examen físico post-natal, como las características faciales, RCIU y alteraciones en las extremidades. Entre los hallazgos descritos en la literatura como ocasionales que se evidenciaron en la autopsia perinatal que incluyó radiografías, estaban la hernia diafragmática, el paladar arqueado y el útero bicorne. Sin embargo, durante las ecografías prenatales se observó la RCIU y no otras alteraciones como el acortamiento de las extremidades y la hernia diafragmática, que podrían haber contribuido a un acercamiento al diagnóstico de SCDL prenatal y enfoque del síndrome en el feto para establecer el pronóstico del embarazo y un plan de trabajo. Dentro de las alteraciones cromosómicas más comúnmente asociadas con RCIU y alteración en las extremidades se encuentran la trisomía 13 y la 18, que se descartaron por el resultado del cariotipo 46, XX.
El diagnóstico diferencial en este caso con RCIU y alteraciones en las extremidades superiores incluye: síndrome de Grebbe, síndrome de Roberts, síndrome de Child y síndrome de TAR, ninguno de los cuales presenta los hallazgos faciales característicos del SCDL como microbraquicefalia, baja implantación del cabello y las orejas, sinofridia, pestańas largas, puente nasal deprimido, narinas antevertidas, labios finos, filtro alargado, boca en carpa, retromicrognatia. La sumatoria de los hallazgos del examen físico, la autopsia, el cariotipo y la historia materna de no haber exposición alguna a tóxicos permite clasificarlo como SCDL tipo I o forma clásica.
El diagnóstico in utero de este síndrome se puede sospechar al encontrar un feto con RCIU y acortamiento severo en las extremidades superiores18. Estos hallazgos deben motivar una búsqueda cuidadosa de otros signos asociados con SCDL como clinodactilia, sindactilia, microbraquicefalia, micrognatia, paladar ojival o hendido, alteraciones cardiovasculares y hernia diafragmática, que no sólo acercan al diagnóstico de SCDL sino que permiten establecer el pronóstico del feto y planear posibles intervenciones in utero o neonatales.
Después de revisar las diferentes series de casos en la literatura y descripciones de textos de dismorfología sobre el SCDL se seleccionaron las características que pueden sugerir el diagnóstico en la ecografía obstétrica o en el recién nacido (Cuadro 1)6-8,16,17. El diagnóstico temprano de esta condición permite iniciar un manejo integral rápido del paciente, que incluye asesoramiento familiar, ayuda médica y estimulación psicomotora apropiada para producir un mejor desempeńo del nińo.
AGRADECIMIENTOS
Agradecemos al grupo ECLAMC Cali por su valiosa ayuda en la búsqueda de recién nacidos con malformaciones congénitas; al personal del Laboratorio de Citogenética del Departamento de Morfología de la Facultad de Salud de la Universidad del Valle por el procesamiento del cariotipo; al Dr. Pedro Rovetto por su orientación en la autopsia del recién nacido; a los estudiantes de Medicina de la Universidad del Valle por su colaboración.
REFERENCIAS
1. Brachmann W. Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnormalitaten (zwerghaftogkeit, halsrippen, behaarung). Jarh Kinder Phys Erzie 1916; 84: 225-235.
2. Lange C. Sur un type nouveau de degenerescence (typus Amstelodamensis). Arch Med Enfants 1933; 36: 713-719.
3. Beck B. Epidemiology of Cornelia de Lange syndrome. Acta Paediatr Scand 1976; 65: 631-638.
4. Martínez-Frías ML, Bermejo E, Félix V, Jiménez N, Gómez-Ullate J, López JA, et al. Síndrome de Brachmann de Lange en nuestro medio: características clínicas y epidemiológicas. An Esp Pediatr 1998; 48: 293-298.
5. Opitz JM. Editorial comment: the Brachmann-de Lange syndrome. Am J Med Genet 1985; 22: 89-102.
6. Ireland M. Cornelia de Lange syndrome. En: Cassidy S, Annanson J (eds.) Management of genetic syndromes. New York: Wiley-Niss; 2001. p. 85-102.
7. Ruiz de la Cuesta-Martín C, Abio-Albero S, García-Bodega O, Rite-Gracia S, López-Pisón J, Vera-Cristóbal F, et al. Síndrome de Cornelia de Lange Neonatal. Rev Neurol 2004; 38: 1027-1031.
8. Jones K, Smith DW. Recognizable patterns of human malformations. 5th ed. Philadelphia: W.B. Saunders Co; 1997. p. 374-375.
9. Bryson Y, Sakati N, Nyhan WL, Fish CH. Self-mutilaty behavior in the Cornelia de Lange syndrome. Am J Med Defic 1971; 76: 319-324.
10. Shear S, Nyhan L, Kirman H, Stern J. Self-mutilative behavioral as a feature of the Lange syndrome. J Pediatr 1971; 78: 506-509.
11. Berney TP, Ireland M, Burn J. Behavioural phenotype of Cornelia de Lange syndrome. Arch Dis Child 1999; 81: 333-336.
12. Smith M, Herrell S, Lusher M, Lako L, Simpson C, Wiestner RS, et al. Genomic organisation of the human chordin gene and mutation screening of candidate Cornelia de Lange syndrome genes. Hum Genet 1999; 105: 104-111.
13. McKusick V, Dolan S. Cornelia de Lange syndrome; CDLS. OMIM. [en línea] 14 julio del 2004. [Fecha de acceso 29 noviembre 2004]. URL disponible en http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=122470
14. Tonkin T, Wang J, Lisgo S, Bamshad J, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nature Genet 2004; 36: 636-641.
15. Van Allen M, Filippi G. Siegel-Bartelt J. Clinical variability within Brachmann-de Lange syndrome: a proposed classification system. Am J Med Genet 1993; 47: 947-958.
16. Jackson L, Kline D, Barr A, Koch S. De Lange syndrome: a clinical review of 310 individuals. Am J Med Genet 1993; 47: 940-946.
17. Ireland M, Donnai D, Burn J. Brachmann de Lange syndrome: delineation of the clinical phenotype. Am J Med Genet 1993; 47: 956-964.
18. Marino T, Wheeler P, Simpson L, Craigo S, Bianchi D. Fetal diaphragmatic hernia and upper limb anomalies suggest Brachmann-de Lange syndrome. Prenat Diagn 2002; 22: 144-147.