Validación del método analítico para la determinación de valsartán en plasma humano por HPLC/UV con adición de estándar empleando losartán como estándar interno
Milena Pérez, Quim. Farm.1, Gloria Ramírez, Quim. Farm.2, Mauricio Pérez, Estad.3, Piedad Restrepo, Quim. Farm. Farmacol.4
1. Directora del Laboratorio Biofarmacéutico, CIDEIM, Cali, Colombia. e-mail: milena_perez@cideim.org.co
2. Asistente de Investigación, CIDEIM, Cali, Colombia. e-mail: gloriashirley_ramirez@cideim.org.co
3. Unidad de Epidemiología y Bioestadística, CIDEIM, Cali, Colombia. e-mail: mauricioperez@cideim.org.co
4. Coordinadora Unidad de Estudios Biofarmacéuticos, CIDEIM, Cali, Colombia.
e-mail: piedad_restrepo@cideim.org.co
Recibido para publicación septiembre 19, 2005 Aceptado para publicación enero 4, 2007
RESUMEN
Introducción: El concepto de validación se refiere a la evaluación estadística de los resultados obtenidos en la aplicación de técnicas analíticas, mediante pruebas convenientemente documentadas y demostrativas de que un método es lo suficientemente fiable a fin de producir el resultado previsto bajo condiciones definidas, como son: sistema analítico, intervalo de concentración, infraestructura y talento humano.
Objetivo: Describir el proceso de validación del método analítico para la cuantificación de valsartán en plasma humano por HPLC-UV y su aplicación en estudios farmacocinéticos, de biodisponibilidad y bioequivalencia de medicamentos que contengan valsartán como principio activo.
Metodología: Se desarrolló un método para la detección y cuantificación de valsartán en plasma humano, con elución isocrática por cromatografía líquida de fase reversa, con detección ultravioleta a 265 nm, mediante el método de adición de estándar. Se utilizó losartán como estándar interno. El método implica una extracción en fase sólida de los principios activos (valsartán y losartán) con cartuchos C8. La separación se realizó en una columna C18 en fase reversa y la fase móvil fue una mezcla de acetonitrilo: buffer fosfato (45:55 v/v) ajustado a pH 2.7±0.1 con ácido fosfórico concentrado. El método se validó en el rango de concentraciones de 0.05 a 20 µg/ml con adición de estándar de valsartán de 2.5 µg/ml.
Resultados y conclusiones: La curva de calibración fue lineal en el rango de concentraciones establecido. Se evaluó la reproducibilidad, estabilidad y porcentaje de recuperación del método. El método para determinar el valsartán en plasma humano por HPLC/UV fue preciso y exacto con un límite de cuantificación de 1.485 µg/ml. Este método fue suficientemente sensible para su aplicación en estudios farmacocinéticos de valsartán.
Palabras clave: Valsartán; Losartán; Extracción en fase sólida; Método de adición de estándar; HPLC/UV.
Validation of an analytical method for the determination of valsartan in human plasma by HPLC/UV with addition standard using losartan as an internal standard
SUMMARY
Introduction: The validation concept refers to the statistical evaluation of the results obtained in the application of analytic technics, by appropriately documented and demonstrative tests that a method is sufficiently reliable to produce the result foreseen under defined conditions, like they are: analytic system, concentration interval, infrastructure and human talent.
Objective: To describe the validation process of the analytic method for the valsartan quantification in human plasma by HPLC-UV and its application in pharmacokinetic, bioavailability and bioequivalence studies of products that contain the active principle valsartan.
Methodology: A method for detection and quantification of valsartan in human plasma has been developed using an isocratic elution on reversed phase liquid chromatography with ultraviolet detection at a single wavelength (265 nm) and the addition standard method. Losartan was used as an internal standard. This method involves a solid-phase drug extraction (valsartan and losartan) from plasma using C8 cartridges. Separation was achieved on a C18 reversed phase column and the mobile phase consisted of 45% acetonitrile and 55% phosphate buffer (adjusted topH 2.7 ± 0.1 with phosphoric acid). The assay has been vali-dated over a concentration range of 0.05 to 20 µg/ml with addition of valsartan 2.5 µg/ml.
Results and conclusions: Calibration curve was linear in the described concentration range. The reproducibility, stability and recovery of the method were evaluated. Determination of valsartan in human plasma by HPLC/UV method was accurate and precise with a quantitation limit of 1.485 µg/ml. The method was sufficiently sensitive for pharmacokinetic studies of valsartan in human plasma.
Keywords: Valsartan; Losartan; Solid-phase extraction; Addition standard method; HPLC/UV.
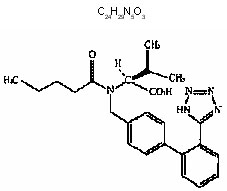
El principio activo valsartán es un antagonista competitivo de la angiotensina II que actúa sobre el subtipo de receptor AT1. El valsartán es un polvo fino blanco o prácticamente blanco; soluble en etanol y metanol y ligeramente soluble en agua. Químicamente se describe como N-(1- oxopentil)-N-[[2’-(1H-tetrazol-5-y1)[1,1’- bifenil]-4-y] metil]-Lvaline. El peso molecular es de 435.5 y su fórmula empírica corresponde a1: Figura 1
La angiotensina II se forma a partir de la angiotensina I en una reacción catalizada por la enzima convertidora de angiotensina (ACE, Kininasa II). La angiotensina es el principal agente vasopresor del sistema renina-angiotensina, con efectos que incluyen vasoconstricción, estímulo y aumento de la síntesis de aldosterona, estímulo cardiaco, y reabsorción renal de sodio. El valsartán bloquea los efectos vasoconstrictores y secretores de aldosterona de la angiotensina II, y bloquea selectivamente la unión de la angiotensina II al receptor AT1 en varios tejidos, tales como el músculo liso vascular y la glándula suprarrenal. Por tanto, su acción es independiente de la vía de síntesis de la angiotensina II1.
El valsartán fue el segundo antagonista competitivo de la angiotensina II que compite sobre el subtipo de receptor AT1 disponible para el tratamiento de la hipertensión. El valsartán se absorbe con rapidez en el tracto gastrointestinal después de su administración oral. Su efecto máximo ocurre dentro de 2 ó 4 horas; y su biodisponibilidad es de 25%. Tiene una vida media de 6 a 9 horas y muestra efectos antihipertensivos por cerca de 24 horas. Alrededor de 9% de las dosis de valsartán se convierten en el metabolito valeril-4-hidroxi-valsartán (M1) inactivo farmacológicamente, mediante biotransformación oxidativa quizá por enzimas del citocromo P450 2C9 y 3A4. La eliminación ocurre sobre todo en la bilis (86%) y en menor cantidad en la orina (13%), principalmente como compuesto inalterado1.
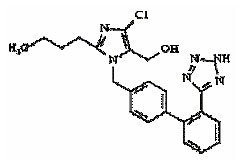
El losartán usado en este método como estándar interno, es un antagonista competitivo de la angiotensina II y agente antihipertensivo. Su fórmula estructural es: Figura 2 .
Hasta el momento, la mayoría de los métodos referenciados para determinar valsartán en el plasma humano utilizan cromatografía líquida con detector de fluorescencia1-3. Este artículo describe un método adecuado para la determinación de valsartán en plasma humano por HPLC/UV para su aplicación en estudios de farmacocinética, biodisponibilidad y bioequivalencia.
MATERIALES Y MÉTODOS
Reactivos. El plasma humano se adquirió comercialmente del Banco de Sangre de la Cruz Roja seccional Valle. El valsartán estándar se obtuvo de la USP. El losartán, que se usó como estándar interno, se adquirió de Chemo. El acetonitrilo (Fisher Scientific) y el agua fueron grado HPLC. El ácido fosfórico (Fisher Scientific), el metanol (Panreac), y el fosfato de potasio monobásico (Merck), fueron grado analítico.
Instrumentación y cromatografía. El estudio se hizo con un equipo de cromatografía líquida LaChrom Elite (Merck-Hitachi) y un detector ultravioleta con arreglo de diodos, Columna Chromolith® Performance RP-18e 100-4.6 mm, 2µm (Merck), software EZChrom Elite 3.1.3 (Merck), balanza analítica (Ohaus Adventurer), agitador vortex (Fisher Scientific), agitador magnético (Multistation IKA), bańo ultrasónico (Fisher Scientific), bańo maría (DIES), purificador de agua (Simplicity Millipore), congelador -70şC (Revco), pH metro (WTW 740 Inolab), cartuchos C8 Sep–Pak (100 mg, 1 ml Waters), centrífuga (Sorvall RC-5B), equipo de extracción en fase sólida (Supelco) y compresor de aire (Atlas Copco).
SECCIÓN EXPERIMENTAL
Preparación del buffer fosfato 0.01M pH 2.0. Se pesaron 1.36 g de fosfato de potasio monobásico, que se diluyeron y disolvieron en 1,000 ml de agua HPLC y se ajustó el pH a 2.0 con ácido fosfórico concentrado.
Preparación del fosfato amortiguado pH 2.7. Se pesaron 1.36 g de fosfato de potasio monobásico, que se diluyeron y disolvieron en 1,000 ml de agua HPLC y se ajustó el pH a 2.7 con ácido fosfórico concentrado.
Preparación de la solución de lavado de cartuchos. Se utilizó una mezcla de buffer fosfato (pH 2.0): metanol (50:50 v/v), como solución de lavado para los cartuchos.
Preparación de la solución de estándar interno. Se preparó una solución de losartán a una concentración de 200 µg/ml, pesando 20 mg de losartán, y llevando a volumen con acetonitrilo en un balón volumétrico de 100 ml.
Preparación del plasma. El plasma humano se conservó almacenado a -70°C en viales de polipropileno de 2 ml, antes de su uso se descongeló a temperatura ambiente.
Preparación de la solución stock de valsartán. Se preparó una solución stock de valsartán a una concentración de 200 µg/ml, pesando 20 mg de valsartán y llevando a volumen con acetonitrilo en un balón volumétrico de 100 ml.
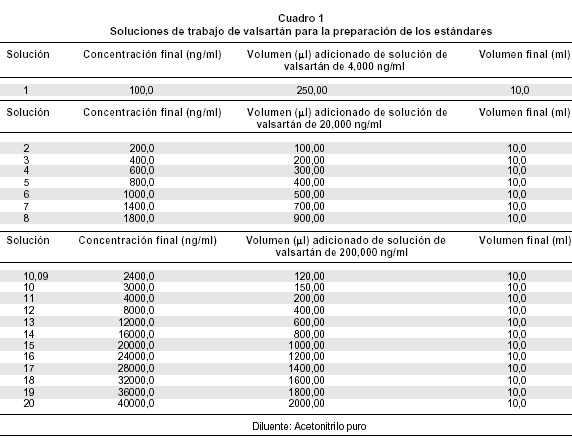
Preparación de las soluciones de trabajo de valsartán. A partir de la solución stock de valsartán (200 µg/ml) se prepararon las soluciones de trabajo como lo muestra el Cuadro 1.
Preparación de la solución de adición de valsartán. Se preparó una solución de valsartán a una concentración de 2.5 µg/ml, transfiriendo 1.25 ml de la solución stock de valsartán (200 µg/ml), a un balón volumétrico de 100 ml y llevando a volumen con acetonitrilo.
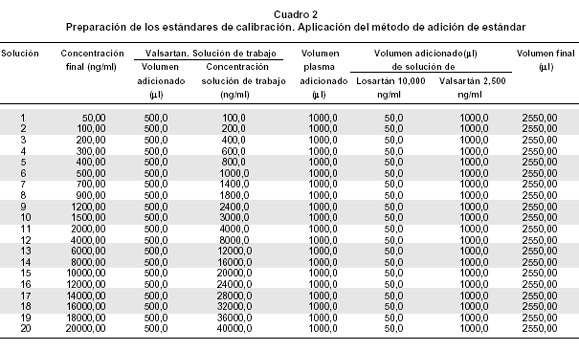
Preparación de las soluciones estándar de calibración. Los estándares de calibración se prepararon tomando los volúmenes descritos en el Cuadro 2, de cada solución de trabajo de valsartán y mezclando con las soluciones descritas en esta.
Acondicionamiento del cartucho. Los cartuchos se acondicionaron con 2 ml de metanol y 1 ml de buffer fosfato pH 2.0. No se debe dejar secar el cartucho.
Preparación de la fase móvil. La fase móvil consistió de una mezcla de acetonitrilo: fosfato amortiguado pH 2.7 (45:55 v/v).
Extracción de los estándares de calibración de valsartán. Cada tubo que contenía una concentración de valsartán, estándar de adición de valsartán 2.5 µg/ml y estándar interno losartán, se agitó y centrifugó por 20 minutos a 6,000 rpm y 4°C. Todo el sobrenadante se transfirió a un cartucho C8 Sep-Pak y se pasó con un vacío menor de 5 mm Hg. El cartucho se lavó con 1 ml de la solución de fosfato amortiguado pH 2.0: metanol (50:50v/v) y se dejó secar con un vacío de 20 mm Hg por 10 minutos. El eluído y el lavado se recogieron en un tubo de ensayo, se agitaron y después se evaporaron a sequedad con aire comprimido en un bańo a 40şC por 1 hora. El residuo fue luego disuelto en 1,000 µl de fase móvil e inyectado (50 µl) en el cromatógrafo.
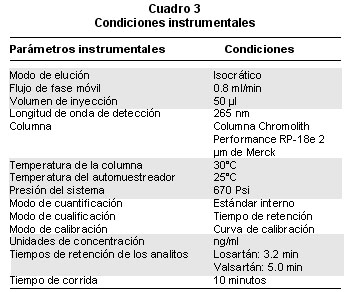
Condiciones cromatográficas. La Gráfica 1 muestra un cromatograma típico de valsartán y losartán obtenido con las condiciones que aparecen en el Cuadro 3.
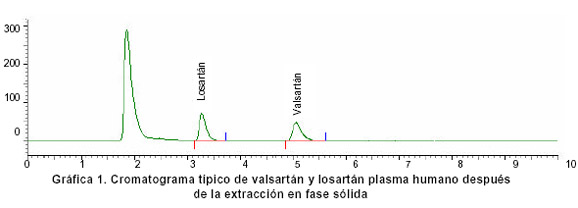
Validación del método de valoración. Para evaluar los parámetros de linealidad, precisión y exactitud del método, se prepararon 20 concentraciones de valsartán, por triplicado en el rango de 0.05 a 20 µg/ml, más la adición de 2.5 µg/ml de valsartán y se determinaron cada una por triplicado. Las muestras se analizaron con la metodología descrita.
Linealidad. La linealidad del método es la relación entre la respuesta del instrumento y las concentraciones conocidas de analito4. La curva de calibración para valsartán se construyó con la relación de áreas de analito a estándar interno versus las concentraciones de analito. Se realizó el análisis de regresión lineal para determinar la linealidad del método así como para generar la ecuación de la curva de calibración: y=mx+b, donde y es la relación de área, x la concentración, m la pendiente y b el intercepto.
Precisión y exactitud. La precisión del método analítico describe la cercanía (grado de dispersión) entre una serie de medidas obtenidas de múltiples repeticiones de una muestra bajo las condiciones establecidas de análisis. La exactitud describe el grado de dispersión del valor obtenido respecto del valor nominal o conocido bajo las condiciones establecidas de análisis5 (Cuadro 4).
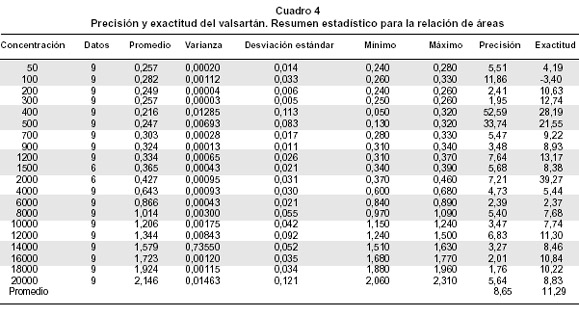
Para el ensayo de precisión intra-día se realizaron 6 inyecciones de una concentración de 2.5 µg/ml de valsartán. La precisión inter-día se hizo al analizar las muestras en dos días distintos.
Recuperación. La recuperación de un analito se refiere a la eficiencia de la extracción en un proceso analítico. Se informa como porcentaje de analito obtenido luego del proceso de extracción5. Para su evaluación, se prepararon por triplicado soluciones de valsartán en plasma a concentraciones de 2.5, 6 y 10 µg/ml, y se analizaron según la metodología descrita. La recuperación (eficiencia de la extracción) se calculó mediante la comparación de las relaciones de área de las muestras extraídas y de los estándares no extraídos.
Especificidad. La selectividad es la capacidad del método bioanalítico de diferenciar el analito en presencia de otros componentes de la muestra. Estos componentes pueden ser metabolitos, impurezas, productos de degradación o componentes de la matriz5. Se analizó la presencia de interferentes cromatográficos como componentes endógenos del plasma mediante 6 muestras de plasma provenientes de distintos donantes, según se sometió antes a la aprobación del comité institucional de ética para investigación en seres humanos.
Sensibilidad. El límite de detección corresponde a la menor cantidad de analito en una muestra que se puede detectar, pero no necesariamente cuantificar con exactitud. El límite de cuantificación corresponde a la menor cantidad de analito en la muestra que se puede cuantificar con precisión y exactitud5. El límite de cuantificación (LOQ) y el límite de detección (LOD) se determinaron experimentalmente a partir de la ecuación de la curva de la linealidad del método por extrapolación de la respuesta a concentración cero. Se tomaron como Y las respuestas promedio y como X las concentraciones.
Estabilidad de los analitos. La estabilidad química de los analitos en el plasma humano se evaluó, al preparar por triplicado tres soluciones de valsartán y losartán en plasma en concentraciones de 2.5, 6 y 10 µg/ml bajo las condiciones específicas que se describen a continuación.
Estabilidad de congelamiento y descongelamiento. Para evaluar la estabilidad de congelamiento y descongelamiento, las soluciones de valsartán en plasma se sometieron a tres ciclos de 24 horas de congelamiento (a -70şC) y descongelamiento. Posteriormente, los resultados obtenidos se compararon con los datos de las muestras frescas.
Estabilidad a corto plazo. Para evaluar la estabilidad a corto plazo, las soluciones de valsartán en plasma se sometieron a descongelamiento a temperatura ambiente durante 4 horas. Después, los resultados obtenidos se compararon con los datos de las muestras frescas.
Estabilidad a largo plazo. Para evaluar la estabilidad a largo plazo, las soluciones de valsartán en plasma se analizaron luego de almacenarlas durante 14 días a -20°C. Los análisis se realizaron al principio y al final del almacenamiento.
Estabilidad post-preparativa. Para evaluar la estabilidad post-preparativa, las soluciones de valsartán en plasma se mantuvieron en el automuestreador a temperatura ambiente (25°C) durante 24 y 90 horas. Después se analizaron y se compararon con el análisis inicial de las muestras frescas.
Estabilidad de las soluciones stock. Para evaluar la estabilidad de las soluciones stock de valsartán y losartán, se prepararon tres soluciones en acetonitrilo y se analizaron después de permanecer 24 horas a temperatura ambiente y una semana a 4°C.
RESULTADOS
Para analizar los resultados, la Unidad de Biometría de CIDEIM utilizó los programas estadísticos SPSS® version 7.5 para Windows, Chicago Inc. 1996 y Statgraphics® Plus for Windows 4.1 Professional version, Copyright 1994-1999 by Statistical Graphics Corp.
Linealidad. Los resultados del modelo lineal describen la relación entre la relación de área y la concentración de valsartán. La ecuación del modelo es:
Relación área = 0.244175 + 0.0000084475 x concentración
Como el valor p de la prueba en la tabla ANOVA es menor de 0.01, hay una relación estadísticamente significativa entre la relación de área y la concentración de valsartán a un nivel de confianza de 99%.
El R-cuadrado estadístico indica que el modelo muestra 99.8% de la variabilidad de la relación de área. El coeficiente de variación igual a 0.9992, indica una fuerte relación entre las variables. El error estándar del estimado muestra una desviación estándar de los residuales de 0.0259329.
Para losartán, el resumen de estadísticas para 177 réplicas muestra un coeficiente de variación de 17.3% e indica la repetibilidad en los resultados cuando se adiciona losartán a 20 concentraciones de valsartán.
Precisión y exactitud. Se obtuvo un coeficiente de variación de 0.3% correspondiente a 6 datos de relación de área de valsartán, lo cual indica que el instrumento es preciso bajo las condiciones de análisis establecidas. Asimismo, la variabilidad en las áreas del estándar interno losartán se determinó sobre 6 datos de los cuales se obtuvo un coeficiente de variación de 1.5%.
La relación F, que en este caso es de 1240.45, es la relación del valor estimado entre grupos y del valor estimado dentro de grupos. El valor p de la prueba F es menor de 0.05, e indica que hay diferencias significativas entre las medias de las relaciones de área de valsartán de cada nivel de concentración, a un nivel de confianza de 95%.
Se obtuvo un coeficiente de variación de 8.65% correspondiente a 20 concentraciones de valsartán, lo cual indica reproducibilidad bajo las condiciones establecidas de análisis.
Se evaluó la precisión inter-día para determinar variaciones en los datos cuando el método lo aplican distintos analistas en días distintos. Los coeficientes de variación de 1.6% y 2.2% para los ensayos 1 y 2 a su vez indican variaciones menores de 30% cuando diversos analistas aplican el método en días diferentes.
Para exactitud, el valor F, que en este caso es de 1228.64 es la relación del valor estimado entre grupos y del valor estimado dentro de grupos. El valor p de la prueba F menor de 0.05, indica que hay diferencias significativas entre las medias de las concentraciones experimentales de valsartán en cada nivel de concentración teórica, a un nivel de confianza de 95%. Se obtuvo un porcentaje de desviación de 11.3% correspondiente a 20 concentraciones de valsartán, y esto seńala exactitud bajo las condiciones establecidas de análisis.
Recuperación. La eficiencia del método de extracción se determinó como porcentaje de recuperación. Se obtuvo una cifra de 90%.
Especificidad. No se encontraron picos que interfirieran de manera significativa con la respuesta de valsartán y losartán.
Estabilidad. Se observan variaciones en la concentración de valsartán cuando se somete a las distintas condiciones de almacenamiento descritas.
Las soluciones stock de valsartán experimentan cambios en su respuesta cuando se conservan a temperatura ambiente (25şC) durante 24 horas y a 4şC durante una semana. La solución stock de losartán no sufre cambios cuando se almacena a las condiciones descritas.
DISCUSIÓN
Un coeficiente de correlación mayor de 0.8000 (0.9992) para la relación de áreas versus concentración, sugiere una fuerte relación entre la respuesta del instrumento y las concentraciones conocidas de valsartán. Un coeficiente de variación menor de 15% (0.4%) indica que el instrumento es preciso bajo las condiciones de análisis establecidas. Un coeficiente de variación promedio menor de 15% (8.6%) muestra que el método analítico es reproducible.
Coeficientes de variación promedio de 1.6% y 2.2%, enseńan que hay una variación menor de 30% en la relación de área cuando el método analítico se aplica de acuerdo con las condiciones de laboratorio, por dos analistas en distintos días.
Un porcentaje de desviación menor de 20% (1.3%) de la concentración experimental con respecto de la concentración real expresa que el método analítico es adecuado para determinar con exactitud diferentes concentraciones de valsartán.
El método analítico con detección ultravioleta proporcionó una adecuada sensibilidad, LOD 445.6 ng/ml y LOQ 1485.13 ng/ml versus lo que informa la literatura3: LOQ 5-60 ng/ml obtenido con detector de fluorescencia.
El método de extracción presenta un porcentaje de recuperación de 90%. No se encontraron picos que interfirieran de manera significativa con las respuestas de valsartán y losartán.
En cuanto a estabilidad se refiere, las muestras de valsartán y losartán en plasma no se deben someter a ciclos de congelamiento y descongelamiento, se deben descongelar a temperatura ambiente y analizar en el menor tiempo posible, y se deben mantener refrigeradas antes de ingresarlas al automuestreador.
La solución stock de valsartán se debe preparar diariamente, mientras que la solución stock de losartán puede permanecer por una semana a 4şC.
CONCLUSIÓN
Se desarrolló un método rápido y específico por HPLC-UV para la determinación de valsartán en muestras de plasma humano, con losartán como estándar interno; el método descrito en este documento presenta ventajas sobre otros previamente informados, en términos de valoración del principio activo por cromatografía líquida con detección ultravioleta y adición de estándar. El método se validó para exactitud, precisión, linealidad y estabilidad. Por tanto, se puede emplear en el análisis de muestras obtenidas en estudios de farmacocinética, biodisponibilidad y bioequivalencia para productos que contengan valsartán como principio activo.
AGRADECIMIENTOS
Los autores agradecen al Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología (COLCIENCIAS) su apoyo financiero al proyecto y al Laboratorio Farmacéutico Tecnoquímicas S.A., por proporcionar los estándares de valsartán y losartán.
REFERENCIAS
1. Center for Drug Evaluation and Research Department of Health and Human Services. Approval Package for Application Number 21-283/S 002. Printed Labeling. Novartis Pharmaceutical Co. Prescribing Information. Approved 2002.
2. Sechaud R, Graf P, Bigler H, Gruendl E, Letzkus M, Merz M. Bioequivalence study of a valsartan tablet and a capsule formulation after single dosing in healthy volunteers using a replicated crossover design. Int J Clin Pharmacol Ther 2002; 40: 35-40.
3. González L, López J, Alonso R, Jiménez RM. Fast screening method for the determination of angiotensin II receptor antagonists in human plasma by high-performance liquid chromatography with fluorimetric detection. J Chromatogr A 2002; 949: 49-60.
4. Daneshtalab N, Lewanczuk RZ, Jamali F. High performance liquid chromotopraphic analysis of angiotensin II receptor antagonist valsartan using a liquid extraction method. J Chromatogr B 2002; 766: 345-349.
5. Food and Drug Administration. Guidance for the industry. Bioanalytical method validation. Food and Drug Administration; 2001.