Fibrosis quística. Aspectos diagnósticos
Luis Ortigosa, M.D.*
* Profesor Asociado de Pediatría, Facultad de Medicina, Universidad de La Laguna, Tenerife, España.
e-mail: lortcas@gobiernodecanarias.org
Recibido para publicación diciembre 28, 2006 Aceptado para publicación enero 25, 2007
RESUMEN
La fibrosis quística (FQ) es una de las enfermedades genéticas mortales más frecuentes en la raza caucásica. Se caracteriza por una disfunción de las glándulas exocrinas, con insuficiencia pancreática y bronconeumopatía crónica. Es una enfermedad de transmisión autonómica recesiva, se sabe que el gen defectuoso está localizado en el cromosoma 7 humano, conocido como gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR),y que de las más de mil mutaciones de este gen, la mutación DF508 es la más común, pues se halla en aproximadamente 70% de los alelos CFTR defectuosos. El diagnóstico de la FQ se ha basado clásicamente en la determinación de por lo menos 2-3 determinaciones positivas de electrólitos en sudor, junto con uno de los siguientes criterios clínicos: íleo meconial, historia familiar de FQ, insuficiencia pancreática exocrina, enfermedad pulmonar crónica, azoospermia obstructiva y síndrome de pérdida de sal. Los criterios diagnósticos actuales incluyen, junto a la presencia de las características clínicas, dos determinaciones de concentraciones de cloro en sudor superior a 60 mmol/l, o demostración de alteraciones en el transporte iónico a través del epitelio nasal (diferencia de potencial nasal) o la detección de dos mutaciones reconocidas de FQ.
Palabras clave: Fibrosis quística; Electrólitos en sudor; CFTR; Mutación DF508; Diferencia de potencial nasal.
Cystic fibrosis. Diagnosis
SUMMARY
Cystic fibrosis (CF) is one of the most frequent inherited mortal diseases in Caucasian population. Dysfunction in exocrine glands is described in CF patients, with severe pancreatic insufficiency and chronic lung disease. CF is inherited as an autosomal recessive disorder. More than 1000 disease-associated mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have been described. DF508 mutation is the most common mutation in the CF gen. Diagnosis in CF is based on clinical and laboratory tests findings. Meconial ileus, CF in other relatives, chronic lung disease, congenital absence of the vas deferens with azoospermia are among other clinical findings, main criteria in CF patients. Two positive results in sweat chloride test , or demonstration in nasal epithelial ionic transport alteration (nasal potential difference) and identification of two CF mutations in the patient are laboratory findings in CF.
Keywords: Cystic fibrosis; Sweat test; Nasal potential difference; CFTR; DF508 mutation.
La fibrosis quística (FQ) o mucoviscidosis, es una de las enfermedades genéticas mortales más frecuentes en la raza caucásica, con una incidencia estimada entre 1 por cada 2,500 a 3,500 recién nacidos vivos. Es una enfermedad de transmisión autosómica recesiva; se calcula que 1 de cada 25 personas es portadora del gen defectuoso de la FQ.
La incidencia de la FQ en las poblaciones no caucásicas es muy baja. A pesar de que los datos no son suficientemente precisos, se evalúa que en la población americana de raza negra, la incidencia es de 1/17,000, mientras que existen pocos casos descritos en población negra africana. En América Latina pocos países han descrito estudios sobre la incidencia real de FQ entre sus poblaciones, aunque existen publicaciones sobre Cuba, México, Costa Rica, Argentina, Brasil, y más recientemente Colombia1.
Es una enfermedad que ataca las células epiteliales exocrinas, las personas afectadas producen un moco espeso y viscoso, que provoca una obstrucción de los conductos de los órganos donde se localiza, siendo el páncreas y los pulmones los órganos más comprometidos, aunque se trata de una entidad multisistémica. De hecho, la enfermedad pulmonar y la insuficiencia pancreática determinan de una forma esencial la evolución, gravedad y mortalidad en la FQ.
En 1936 Fanconi et al.2 hicieron la primera descripción de la FQ y asociaron la fibrosis congénita del páncreas con la presencia de bronquiectasias.
En 1934 Andersen3 describe los hallazgos clínicos y anatomopatológicos de los enfermos con FQ, y denominó por primera vez con el nombre de fibrosis quística del páncreas a esta enfermedad, por los quistes y fibrosis del tejido pancreático que presentan estos enfermos.
Una década después, Farber4 propuso el término de «mucoviscidosis», por considerar que la hiperviscosidad del moco era la causa fundamental de las alteraciones de los diversos órganos que presentaban los enfermos de FQ, y que la pérdida de función y fibrosis observadas eran hechos secundarios al bloqueo de los conductos excretores por las secreciones espesas.
Prácticamente hasta 1953 el diagnóstico de FQ se hacía con base en datos clínicos, ante la observación de neumopatía crónica + insuficiencia pancreática, pues no se contaba con herramientas de laboratorio que apoyasen la sospecha clínica de FQ. En ese ańo, Di Sant´Agnese et al.5 demostraron que la causa del choque hiponatrémico que presentaban muchos de estos pacientes durante la ola de calor que sufrió la ciudad de Nueva York, se debía a la elevada concentración anormal de cloro eliminada por el sudor; posteriormente este descubrimiento se constituyó en la prueba diagnóstica más importante de apoyo a la sospecha clínica de la enfermedad (prueba del sudor), mediante la determinación de electrólitos en sudor obtenido por iontoforesis con pilocarpina, prueba desarrollada por primera vez por Gibson y Cooke en 19595.
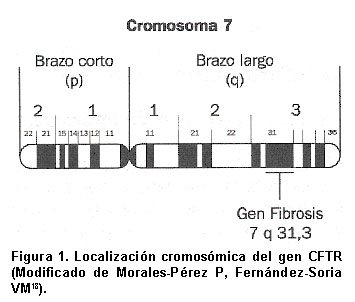
Y para finalizar este breve recorrido por la historia de la FQ, hay que seńalar la importancia del descubrimiento del gen de la FQ: en 1985 se identificó y localizó la mutación responsable de la FQ en el brazo largo del cromosoma 7 humano7, concretamente en la región 7q31-32, y cuatro ańos más tarde, en agosto de 1989, en un esfuerzo común entre dos grupos de investigadores, coordinados por Tsui en Toronto y Collins en Michigan, se comunicó el descubrimiento del gen responsable de la enfermedad8-10 analizando la secuencia de cADN normal y de pacientes con FQ, demostrando al mismo tiempo que la mutación más común es la delección de tres pares de bases que codifican un único aminoácido (fenilalanina), mutación DF508, aunque hoy se sabe que existen más de mil mutaciones para este gen11.
A mediados de los ańos 30, cuando se describió por primera vez la FQ, la esperanza de vida para los nińos con esta grave enfermedad no superaba los primeros meses o ańos de vida, pero esta circunstancia está cambiando radicalmente, gracias a los importantes avances en el conocimiento de las bases genéticas y fisiopatológicas de la FQ producidos en las últimas décadas del siglo XX y comienzo del XXI, al diagnóstico cada vez más precoz de la enfermedad, y a la puesta en funcionamiento de unidades monográficas de referencia de FQ, donde los pacientes de FQ se tratan de forma multidisciplinaria, con una notable mejoría en la calidad y esperanza de vida, superior a los 30 ańos en los lugares que cuentan con estos centros especializados en el seguimiento y manejo de enfermos con FQ12,13.
ASPECTOS GENÉTICOS Y FISIOPATOLÓGICOS
El gen de la fibrosis quística (CFTR). Desde principios de 1980, varios grupos de investigadores iniciaron estudios de ligamiento genético entre la FQ y distintos marcadores de DNA, con objeto de identificar el gen de la FQ. A finales de 1985, Tsui et al.7 describieron el liga-miento genético entre la FQ y el marcador ADN DOCRI-917, asignado luego al cromosoma 7, estudios que se ampliaron en ańos posteriores, descubriéndose nuevos marcadores (MET y D7S8, ...) ligados al gen FQ, que permitieron la construcción de un mapa de ligamiento genético entre estos marcadores y la enfermedad por otros grupos de investigadores en distintas partes del mundo entre los ańos 1985 y 198914-17, de forma que el análisis de fragmentos de restricción polimórficos (RPLF) durante esos cuatro ańos permitió determinar la existencia de un único locus, el gen de la FQ, en el brazo largo del cromosoma 7 humano, concretamente en la región q31 (Figura 1).
En agosto de 1989, en un estudio coordinado por Tsui en Toronto y por Collins en Michigan8-10, se clonó el ADN comprendido en esta región, con estrategias de walking y jumping cromosómico para llegar a secuenciar el cADN del gen de la FQ, al que se denominó CFTR (regulador de la conductancia transmembrana de la FQ), al mismo tiempo que se logró caracterizar la mutación más frecuente de este gen, la mutación DF508.
El gen de la FQ fue el primer gen humano identificado sin conocer la proteína para la que codificaba, ni disponer de claves citogenéticas que permitiesen un avance rápido en su identificación18, mediante la tecnología denominada «genética reversa», que consiste en la identificación y aislamiento de un gen determinado, sin tener datos sobre la función y características bioquímicas de la proteína para la que éste codifica.
El gen CFTR tiene un tamańo de 250 kilobases, comprende 27 exones y codifica para una proteína de 1,480 aminoácidos, que se localiza en la membrana apical de las células epiteliales normales. Esta proteína pertenece a una «superfamilia» de glucoproteínas P de transporte de membrana, y actúa como un canal iónico de cloro, regulador del transporte iónico a través de la membrana apical de las células epiteliales, caracterizada por la falta de respuesta a AMPc19.
Es una proteína constituida por dos regiones transmembránicas (hidrofóbicas) separadas por una región de unión al ATP (región NBF, nucleotide binding fold) y una subunidad reguladora R (hidrófila), seguidas de otra región NBF (Figura 2).
Mutaciones en el gen CFTR. Las mutaciones en el gen de la FQ se distribuyen a lo largo de todo el gen, aunque se producen con mayor frecuencia en algunas regiones del mismo, y presentan considerables variaciones según el grupo étnico y/o el área geográfica estudiad1,14,19,20.
La mutación DF508 es la más frecuente; se han descrito en la actualidad más de 1,300 mutaciones del gen CFTR11. La identificación del gen y de sus mutaciones ha permitido el estudio de la correlación fenotipo/genotipo mediante el análisis de la relación de las diferentes mutaciones presentes con la expresión clínica de la enfermedad, tal como se refleja a continuación19:
Se han sugerido cinco mecanismos moleculares por los que una mutación produciría una disfunción del canal CFTR: las mutaciones clase I y II suelen asociarse con insuficiencia pancreática (las mutaciones de clase I conducen a una ausencia en la producción de CFTR, y las de clase II no permiten que la proteína alcance la membrana epitelial), mientras que las otras tres clases de mutaciones (III, IV y V) presentan una gran variabilidad en su expresión clínica21-23.
ASPECTOS FISIOPATOLÓGICOS
Los estudios fisiopatológicos en la FQ, fundamentalmente durante las décadas de 1980 y 1990, unidos a los avances en el conocimiento de las alteraciones genéticas que se comentaron antes, han permitido grandes avances en el conocimiento del defecto básico de la FQ: éste consiste en una alteración de la regulación del canal de cloro activado por AMP cíclico, que se encuentra en la membrana apical de las células epiteliales secretoras14,24,25.
La proteína CFTR no funciona adecuadamente a nivel de la membrana celular apical, ni como canal de cloro, ni como regulador de los canales rectificadores exteriores del cloro y de los canales de sodio, siendo ésta la primera y principal alteración/disfunción que genera la enfermedad , y que explica porqué los enfermos con FQ tienen el sudor tan salado, porqué necesitan enzimas pancreáticas cuando presentan insuficiencia pancreática, porqué se infectan crónicamente sus pulmones y porqué pueden desarrollar cirrosis biliar y diabetes mellitus24, entre otras complicaciones evolutivas.
ASPECTOS DIAGNÓSTICOS
El diagnóstico de la FQ se ha basado clásicamente en la determinación de por lo menos 3 determinaciones positivas de electrólitos en sudor, junto con uno de los siguientes criterios clínicos:
Pero una vez conocido el gen defectuoso de la FQ y la identificación de sus mutaciones, se han publicado diferentes casos de enfermos de FQ con ciertas mutaciones infrecuentes, que pueden estar asociados a una prueba del sudor normal25, o expresiones clínicas atípicas de FQ (como agenesia de vasos deferentes, sin compromiso pancreático ni pulmonar)21, por lo que recientemente se ha propuesto que para efectuar el diagnóstico de FQ sea necesario por lo menos uno de los criterios clínicos seńalados, junto a la determinación patológica de electrólitos en sudor o bien la constatación de 2 mutaciones en el gen FQ26,27, o diferencia de potencial nasal anormal.
Determinación de electrólitos en sudor. La determinación de electrólitos en el sudor, o prueba de sudor, es la principal herramienta para el adecuado diagnóstico de la FQ desde la publicación de Gibson y Cooke5 del método de iontoforesis con pilocarpina (este método recibió el nombre de QPIT: quantitative pilocarpine iontophoretic test).
La prueba del sudor consta de tres etapas: estimular la sudoración, recoger el sudor y por último, determinar la concentración de electrólitos26.
El resultado de la prueba se considera:
Para efectuar esta técnica se precisa de personal experto y habituado a practicarla, porque errores en su realización pueden dar lugar a resultados falsamente positivos o negativos (Cuadros 1 y 2).


Con respecto a los métodos, el desarrollado por Gibson y Cooke es el que tiene mayor reproductibilidad y especificidad. Recientemente tan sólo se admiten dos métodos como válidos: el original de Gibson y Cooke6, QPIT, y el Macroduct28. Dado lo engorroso de la técnica de recoger el sudor por el método original de Gibson y Cooke, posteriormente se han introducido otros sistemas más prácticos, como la recolección del sudor en cubeta de plástico, o más recientemente el sistema Wescor Macroduct, en el que el sudor se recoge en un disco cóncavo de plástico, conectado a un tubo espiral de plástico, y se eliminan así los problemas de evaporación y condensación inherentes al sistema de la cubeta; en la actualidad se reconoce como un método de gran fiabilidad y reproductibilidad, pero tiene el inconveniente del alto costo del material fungible26.
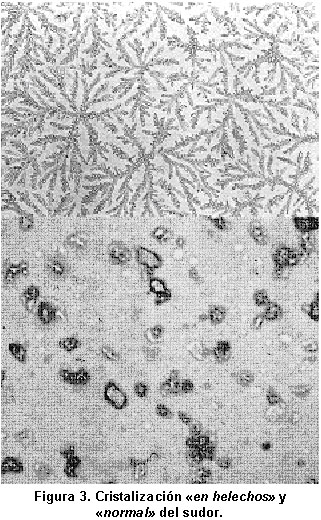
También se debe citar la prueba de desecación o cristalización del sudor, método que puede servir de gran ayuda en el diagnóstico de FQ, en aquellos casos en los que la determinación de electrólitos en sudor da valores dudosos (Cl- entre 50 y 60 mEq/l), y existe sospecha clínica de FQ.
Este método, descrito por primera vez por Kopito et al.29 en 1976 y en el que distintos grupos de investigadores hemos ido adquiriendo experiencia, con resultados positivos30,31, se basa en las formas de desecación del sudor en enfermos con FQ, que adopta formas en figuras de «helecho» o «dendritas» muy características, en contraposición con las formas «cuboides» típicas de las personas no FQ. Este es un método complementario a la determinación de electrólitos en el sudor, muy sencillo y económico de realizar en unidades de FQ con pocos medios, pero nunca debe utilizarse como método diagnóstico único, sin determinar Cl- y Na+ en sudor (Figura 3).
Indicaciones clínicas para efectuar la prueba del sudor. Hay una serie de circunstancias clínicas que hacen sospechar la posibilidad de estar ante un paciente con FQ, en las cuales se imdica la determinación de electrólitos en el sudor, y que se han resumido en situaciones de tipo respiratorio (Cuadro 3), digestivo (Cuadro 4) y otras causas diversas (Cuadro 5).



Diferencia de potencial nasal. La diferencia de potencial nasal es una técnica que no está al alcance de todos los laboratorios, por su costo y dificultad técnica, por lo que a pesar de su alta fiabilidad, prácticamente se realiza exclusivamente en centros de investigación o a aquellos pacientes en los cuales no es posible realizar la iontoforesis para determinar los electrólitos en sudor32-34.
La diferencia de voltaje entre la mucosa nasal y la lectura de un electrodo en el antebrazo se correlaciona con el movimiento de sodio a través de las membranas, presentando los pacientes con FQ una diferencia de potencial nasal más negativo que los individuos sanos, siendo los valores normales entre 1.8-53 mV.
Como se comentó, esta técnica no es fácil de efectuar, requiere personal calificado para su realización, consume mucho tiempo y se deben hacer controles frecuentes de calidad, incluida la determinación de por lo menos un control normal el mismo día en que se vaya a practicar la prueba. Además, es necesario establecer valores de referencia para cada laboratorio.
Diagnóstico molecular. El diagnóstico molecular constituye un elemento indispensable para prevenir las enfermedades genéticas hereditarias. En el caso de la FQ, esta prevención es factible mediante la identificación de portadores, el diagnóstico prenatal y más recientemente el diagnóstico preimplantacional. Además, la estrategia actual permite el estudio rápido y eficaz de otras enfermedades relacionadas con la FQ. Por último, los estudios de correlación genotipo-fenotipo contribuyen a la búsqueda de otros genes implicados en la modulación del gen CFTR22.
Diagnóstico basado en los aspectos clínicos. La presentación clínica de la FQ es muy variable, pues se manifiesta habitualmente con insuficiencia pancreática exocrina en 80% a 90% de los casos, y trastornos respiratorios, que son los que van a condicionar el pronóstico y evolución de la enfermedad (las primeras infecciones suelen ser por Haemophilus influenzae y Staphyloccocus aureus), asociándose tras un período variable con infección por Pseudomonas aeruginosa, infección que va a condicionar la evolución de la enfermedad hacia la insuficiencia respiratoria progresiva.
Aproximadamente entre 50% y 75% de los casos de FQ se diagnostican en los primeros ańos de vida del nińo, variando el tipo de presentación clínica en relación con la edad y curso evolutivo natural que haya seguido la enfermedad hasta el momento de realizarse el diagnóstico, como puede apreciarse en los (Cuadros 6, 7 y 8).



La FQ se manifiesta de modo habitual con insuficiencia pancreática, enfermedad pulmonar progresiva, alteración en la secreción de electrólitos en sudor, azoospermia en los varones y disminución de la fertilidad en el sexo femenino. La mayoría de los pacientes con la forma clínica clásica de presentación de la enfermedad (diarrea crónica con insuficiencia pancreática, con o sin compromiso pulmonar inicial y prueba del sudor salado), se diagnostican a lo largo del primer ańo de vida (Cuadro 6.
La afección pulmonar, que constituye la causa más frecuente de mortalidad en estos enfermos, presenta una gran heterogeneidad, tanto en la edad de comienzo, como en el curso evolutivo (Cuadros 7 y 8).
Ya a partir de la adolescencia y en la edad adulta, la enfermedad pulmonar crónica, con infecciones pulmonares severas, y la participación de otros órganos digestivos (fundamentalmente el compromiso hepático y la participación del páncreas exocrino, con diabetes mellitus en 20% y 30% de los casos) va a marcar el pronóstico de la enfermedad (Cuadro 8)
Para el seguimiento clínico de los pacientes con FQ se han desarrollado unos sistemas de evaluación clínica, conocidos como scores de evaluación clínica, o de seguimiento clínico36, siendo el más conocido y utilizado por las unidades de FQ, el score de Shwachman y Kulczycki36, modificado por Doershuk et al.38. Este score consta de 4 puntos, con una puntuación máxima de 25 puntos en cada uno de ellos:
La puntuación ideal es 100 y el estado de los enfermos se clasifica así:
Este puntaje, a pesar de los ańos transcurridos desde su publicación, aún es de gran utilidad para evaluar el estado clínico de los enfermos con FQ, aunque no incluye datos de función pulmonar, pero es muy fácil de efectuar en unidades de FQ con pocos medios técnicos, ya que sólo precisa de una radiografía de tórax y datos clínicos de fácil evaluación por médicos expertos en el seguimiento de estos pacientes.
Para completar los puntajes clínicos con el grado de compromiso pulmonar se han propuesto distintos scores basados en técnicas de diagnóstico por la imagen, siendo las más conocidas y utilizadas los scores basados en radiografía de tórax por Chrispín y Norman39 o la modificación realizada al mismo por Brasfield et al.40
Estos scores radiológicos valoran los siguientes parámetros:
En los últimos ańos, con el advenimiento de nuevos métodos de diagnóstico por la imagen, se han propuesto nuevos scores basados en los hallazgos de tomografía axial computadorizada (TAC) de alta resolución41 o mediante el empleo de gammagrafía pulmonar de ventilación42, métodos que, aunque encuentran una excelente correlación con las pruebas de función pulmonar, son de dudosa utilidad en el seguimiento ordinario de los pacientes con FQ.
CONCLUSIONES
Hasta mediados del siglo pasado, los nińos enfermos de FQ morían en los primeros ańos de su vida, debido a una combinación fatal del cuadro de malabsorción inpruebainal con malnutrición y la enfermedad pulmonar grave. Gracias a los avances en la investigación del defecto básico de la enfermedad, y al mejor conocimiento de la fisiopatología y mecanismos etiopatogénicos de la FQ (conseguido fundamentalmente en las últimas décadas), con importantes y novedosos logros en los métodos de diagnóstico y en el manejo y tratamiento de estos pacientes43-46, se alcanzan mejorías en la calidad y esperanza de vida para estos pacientes, inimaginables hace tan sólo treinta ańos.
REFERENCIAS
1. Jay LM, Mateus H, Fonseca D, Restrepo CM, Keyeux G. PCR-heterodúplex por agrupamiento: Implementación de un método de identificación de portadores de la mutación más común causal de fibrosis quística en Colombia. Colomb Med 2006; 37: 176-182.
2. Fanconi G, Vehlinger E, Knauer C. Das Coeliakie-Syndrom bei angeborenem zystischem Pancreas fibromatose und Bronchiek-tasien. Wien Med Wschr 1936; 86: 753-756.
3. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathological study. Am J Dis Child 1938; 56: 344-349.
4. Farber S. Pancreatic function and disease in early life. V. Pathologic changes associated with pancreatic insufficiency in early life. Arch Pathol 1944; 37: 238-243.
5. Di Sant´ Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas, its clinical significance and relationship to the disease. Pediatrics 1953; 12: 549-563.
6. Gibson LE, Cooke RE. A test for concentration of electrolytes in cystic fibrosis utilizing pilocarpine by iontophoresis. Pediatrics 1959; 23: 545-563.
7. Tsui LC, Buchwald M, Barker D, Braman JC, Knowlton R, Schrumm JW, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 1985; 23: 1054-1057.
8. Rommens J, Iannuzzi M, Kerem B, Drumm M, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: Chromosome Walking and Jumping. Science 1989; 245: 1059-1065.
9. Riordan J, Rommens J, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066-1073.
10. Kerem B, Rommens J, Buchanam J, Markewicz D, Cox T, Chakrauarti A, et al. Identification of the cystic fibrosis gene: Genetics analysis. Science 1989; 245: 1074-1080.
11. Cystic Fibrosis Genetic Analysis Consortium. [falta fecha de acceso]. URL disponible en http://www.genet.sickkids.on.ca/cftr/app
12. Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax 1991; 46: 881-885.
13. Salcedo-Posadas A, Costilla-García A. Organización y funcionamiento de unidades de fibrosis quística. En: Salcedo-Posadas A y García-Novo A (eds). Fibrosis quística. Madrid: PC Works SL; 1997. p. 401-406.
14.Estivill X. Genética molecular de la fibrosis quística. An Nestle 1992; 49: 30-47.
15. White R, Woodward S, Leppert M. A closely linked marker for cystic fibrosis. Nature 1986; 318: 382-384.
16. Estivill X, Schmidtke J, Williamson R, Wainwright BJ. Chromosome assignment and restriction fragment length polymorphism analysis of the anonymus DNA probe B79a at 7q22 (HGM8 assignment D7SS13). Hum Genet 1986; 74: 320-322.
17. Estivill X, McLean C, Nunes V. Isolation of a new DNA marker in linkage desequilibrium with cystic fibrosis situated between J3.11 (D7S8) and IRP. Am J Hum Genet 1989; 44: 704-710.
18. Morales-Pérez P, Fernández-Soria VM. Identificación, estructura y expresión del gen CFTR. En: Salcedo-Posadas A y García-Novo A (eds). Fibrosis quística. Madrid: PC Works SL; 1997. p. 11-26.
19. Antińolo G, Chillón M, Sánchez J. Genética de la fibrosis quística. En: Dapena Fernández FJ (ed.). Fibrosis quística. Atención integral, manejo clínico y puesta al día. Granada: Editorial Alhulia Salobreńa; 1998. p. 41-82.
20. Nuńes-Martínez V, Casals-Senent T. Análisis de las mutaciones del gen. Relación fenotipo/genotipo. En: Salcedo-Posadas A y García-Novo A (eds). Fibrosis quística. Madrid: PC Works SL; 1997. p. 35-44.
21. Casals T, Bassas L, Egozcue S, Ramos MD, Giménez J, Segura A, et al. Heterogeneity for mutations in the CFTR gene and clinical correlations in patients with congenital absence of the vas deferens. Hum Reprod 2000; 15: 1476-1483.
22. Casals-Senent T. El gen CFTR: Diagnóstico molecular en una población heterogénea. An Esp Ped 1999; 133 (Suppl): 37.
23. Estivill X, Ortigosa L, Pérez-Frías J, Dapena J, Ferrer J, Peńa J, et al. Clinical characteristics of 16 cystic fibrosis patients with the missense mutation R334W, a pancreatic insufficiency mutation with variable age of onset and interfamilial clinical differences. Hum Genet 1995; 95: 331-336.
24. Dapena-Fernández FJ, Ramos-Mayo C. Fisiopatología y patogenia de la fibrosis quística. En: Dapena Fernández FJ (ed.). Fibrosis quística. Atención integral, manejo clínico y puesta al día. Granada: Editorial Alhulia Salobreńa; 1998. p. 83-104.
25. Highsmith NE, Burch LH, Zhou Z, Olsen JC, Boat TE, Spock A, et al. A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N Engl J Med 1994; 331: 974-980.
26. Vázquez C. La prueba del sudor en el diagnóstico de la fibrosis quística. En: Dapena Fernández FJ (ed.). Fibrosis quística. Atención integral, manejo clínico y puesta al día. Granada: Editorial Alhulia Salobreńa; 1998. p. 311-324.
27. Kerem E, Kerem B. Genoyipe-phenotype correlations in cystic fibrosis. Pediatr Pulmol 1996; 22: 387-395.
28. Webster HI, Barlow AX. New approach to cystic fibrosis diagnosis by use of an improved sweat induction/collection system and osmometry. Clin Chem 1981; 27: 385-387.
29. Kopito L, Ploss RS, Shwachman H. Crystal forms in sweat from patients with cystic fibrosis. Mod Probl Pediatr 1976; 10: 203-206.
30. Ferrer-Calvete J, Ribes C, Montero C. The sweat crystallization test in the diagnosis of cystic fibrosis. J Pediatr Gastrenterol Nutr 1990; 10: 416-417.
31. Ortigosa L, Oliva C, Duque R, Muro M, León C, Martín L, et al.Prueba de cristalización del sudor y fibrosis quística. II Congreso Nacional de Fibrosis Quística. Barcelona 22-23 noviembre, 1991. Libro de Comunicaciones. p. 102.
32. Jaramillo MA, Vera JF, Briceńo GD, Suárez MA, Barón O. Fibrosis quística: Aspectos gastroinpruebainales y nutricionales. En: Vera-Chamorro JF, Suárez MA, Briceńo GD (eds.). Guía de gastrohepatología y nutrición pediátrica basadas en la evidencia. Bogotá: Distribuna Editorial Médica; 2006. p. 209-262.
33. Escobar H, Kirschlagger E. Fibrosis quística. En: Tratamiento en gastroenterología, hepatología y nutrición pediátrica. Madrid: Sociedad Espańola de Gastroenterología, Hepatología y Nutrición Pediátrica; 2000. p. 213-222.
34. García-Hernández N. Métodos diagnósticos. En: Salcedo-Posadas A y García-Novo A (eds). Fibrosis quística. Madrid: PC Works SL; 1997. p. 273-280.
35. Ferrer-Calvete J. Fibrosis quística. Madrid: Kalifarma ed; 1991. p. 5-26.
36. Sánchez-Solís de Querol M, Pajarón de Ahumada M. Scores de seguimiento. En: Salcedo-Posadas A y García-Novo A (eds). Fibrosis quística. Madrid: PC Works SL; 1997. p. 315-328.
37. Shwachmann H, Kulczycki L. Long-term study of 105 patients with cystic fibrosis: Studies made over a 5 to 14 years period. Am J Dis Child 1958; 96: 6-15.
38. Doershuk CF, Matthews LW, Tucker AS. A 5 years clinical evaluation of a therapeutic program for patients with cystic fibrosis. J Pediatr 1964; 65: 677-693.
39. Chrispin AR, Norman AP. The systematic evaluation of the chest radiograph in cystic fibrosis. Pediatr Radiol 1974; 2: 101-106.
40. Brasfield D, Hicks G, Soong S, Tiller R. The chest roentgenogram in cystic fibrosis: a new scoring system. Pediatrics 1979; 63: 24-29.
41. Nathanson I, Conboy K, Murphy S, Afshani E, Kuhn JP. Ultrafast computerized tomography of the chest in cystic fibrosis: a new scoring system. Ped Pulmonol 1991; 11: 81-86.
42. Kuni CC, Budd JR, Regelman WE, Cucret RP, Boudreau RJ. Comparison of Tc-99m DTPA aerosol ventilation studies with pulmonary function testing in cystic fibrosis. Cl Nucl Med 1993; 18: 15-18.
43. Borowitz D, Durie PR, Clarke LL, Werlin SL, Taylor CJ, Semler J, et al. Gastrointestinal outcomes and confounders in cystic fibrosis. J Ped Gastroenterol N 2005; 41: 273-285.
44. Orenstein DM, Winnie GB, Altman H. Cystic fibrosis: a 2002 update. J Pediatr 2002; 140: 156-164.
45. Rowe SM, Miller A, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352: 1992-2001.
46. Kabra SK, Kabra M, Shastri S, Lodha R. Diagnosing and managing cystic fibrosis in the developing world. Paediatr Respir Rev 2006; 7: 147-150.