Apoptosis de fibroblastos gingivales en periodontitis
Roger Mauricio Arce, O.D.,M.Sc.1, Oscar Tamayo, B.Sc, M.Sc.2, Armando Cortés, M.D.3
1. Profesor Auxiliar, Escuela de Odontología, Facultad de Salud, Universidad del Valle, Cali, Colombia. e-mail: roger_arce@dentistry.unc.edu
2. Profesor Auxiliar, Facultad de Medicina, Universidad Santiago de Cali, Cali, Colombia. e-mail: oscartam@univalle.edu.co
3. Profesor Titular y Jefe del Departamento de Patología, Escuela de Medicina, Facultad de Salud, Universidad del Valle, Cali, Colombia. e-mail: acortes59@gmail.com
Recibido para publicación junio 7, 2006 Aceptado para publicación julio 4, 2007
RESUMEN
Introducción: Los fibroblastos gingivales humanos (FGH) tienen un papel importante en la enfermedad periodontal, pues alteran su normal funcionamiento en respuesta a estímulos pro-inflamatorios. Se cree que los fibroblastos se pueden eliminar anormalmente por medio de apoptosis en periodontitis. El propósito de este estudio es determinar y cuantificar la apoptosis de FGH en biopsias del periodonto de individuos sanos y con enfermedad periodontal.
Métodos: Se realizó un estudio clínico descriptivo de corte transversal en personas con diagnóstico de salud periodontal (S), gingivitis (G) y periodontitis crónica (PC). Se tomaron biopsias escisionales y se hicieron tinciones inmunohistoquímicas (hematoxilina-eosina, caspasa-3 y vimentina). Las placas se interpretaron por histopatología y se digitalizaron para cuantificar las células apoptóticas. Todos los datos se analizaron con un software estadístico para encontrar diferencias significativas (p<0.05).
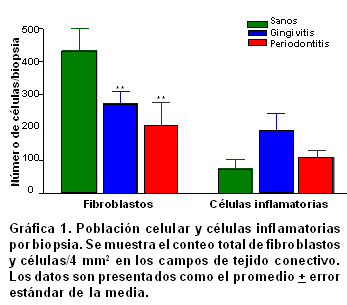
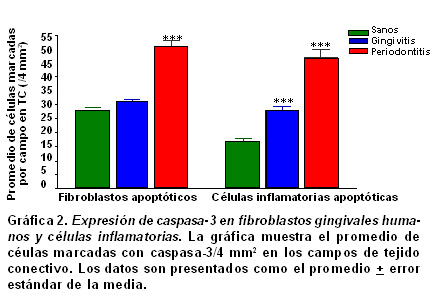
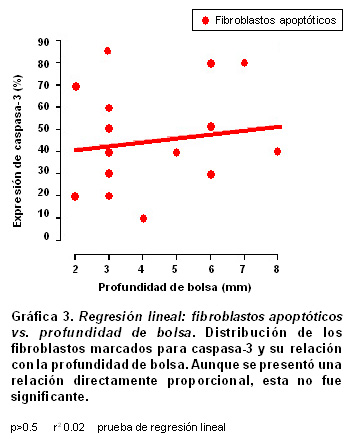
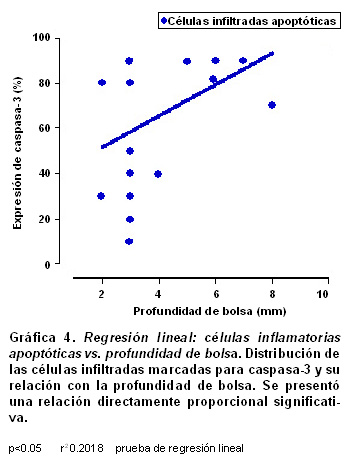
Resultados: La población celular total de fibroblastos tuvo un promedio de 430±67.6 en los individuos sanos y una disminución significativamente progresiva en gingivitis (270±37.1) y periodontitis crónica (206.5±69.8) (p<0.05). La expresión de fibroblastos apoptóticos por campo aumentó de acuerdo con las severidades de la enfermedad [28±16 en sanos (6.5%); 31±17 en gingivitis (11.5%) y 51±24 en periodontitis (24.8%.), (p<0.001)]. La relación entre la expresión de fibroblastos apoptóticos y la profundidad de la bolsa periodontal no fue significante (p>0.5, r²=0.02); mientras que para las células inflamatorias se encontró una relación proporcional significativa (p<0.05, r²=0.2018).
Conclusiones: Los resultados permiten concluir que tanto los fibroblastos gingivales como las células inflamatorias presentan apoptosis manifiesta por la expresión de caspasa-3, y ésta se incrementa significativamente en gingivitis y enfermedad periodontal.
Palabras clave: Periodontitis; Apoptosis; Gingivitis; Patogénesis; Inmunohistoquímica.
Apoptosis of gingival fibroblasts in periodontitis
SUMMARY
Introduction: Human gingival fibroblasts (HGF) have an important role in the periodontal immune response. The fibroblasts alter their normal behavior in response to pro-inflammatory cytokines. It is believed that HGF can be diminished and/or eliminated by means of apoptosis. The purpose of this study was to determine and to quantify apoptosis of HGF in periodontium biopsies from healthy and chronic periodontitis patients.
Methods: A clinical cross-sectional study in people with healthy periodontium (S), gingivitis (G) and chronic periodontitis (PC) patients was carried out. The periodontal biopsies were obtained and immunostained by means of: hematoxylin-eosin, caspase-3, vimentin and caspase-vimentin double-staining for specific visualization of apoptotic fibroblasts. Histopathological and digital analyses were performed. Descriptive statistics were applied to categorical and nominal variables.
Results: Total cell population of HGF had an average of 430±67.6 cells/field in healthy people, and a significantly progressive decrease in gingivitis (270±37.1) and chronic periodontitis groups (206.5±69.8) (p<0.05). As for total population of inflammatory cells, an increase was noticed in gingivitis (191.8±50.1) and a decrease in periodontitis (109.3±21.7) without statistical significance. The expression of apoptotic HGF per field increased accordingly to the severity of the disease [28±16 in health (6.5%); 31±17 in gingivitis (11.5%) and 51±24 in periodontitis (24.8%), p<0.001]. Similar findings were observed for inflammatory cells with different percentage expression [17±13 in health (23%); 28±19 in gingivitis (14.6%) and 47±35 in periodontitis (43.1%), p<0.05]. The relationship between the percentage of expression of apoptotic cells and probing pocket depth was proportional but not significant (p>0.5, r²=0.02); while for the inflammatory cells a significant relationship was observed (p<0.05, r²=0.2018).
Conclusions: The results in this report conclude that HGF and inflammatory cells show apoptosis (caspase-3 expression), and apoptotic cells are significantly increased in gingivitis and chronic periodontitis groups.
Keywords: Periodontitis; Gingivitis; Apoptosis; Pathogenesis; Gingival; Fibroblasts; Immunohistochemistry.
La enfermedad periodontal o periodontitis representa la expresión clínica de una serie de entidades patológicas que tienen en común la destrucción inflamatoria de los tejidos de soporte del diente, incluso el tejido conectivo gingival, el ligamento periodontal y el hueso alveolar. La gingivitis es la inflamación de los tejidos blandos alrededor del diente sin destrucción resortiva del periodonto. Clínicamente la gingivitis se caracteriza por algunos cambios tisulares en la gingiva como enrojecimiento, edema, engrosamiento, ulceración y sangrado. En la enfermedad periodontal se comparten estas características clínicas, con la diferencia principal de la presencia de la bolsa periodontal, resultado de la profundización patológica del surco gingival a causa de la migración apical del epitelio de unión.
La etiología infecciosa de esta enfermedad se reconoció desde la década de 19601. A partir de entonces, los estudios indicaron que la periodontitis representa una infección polimicrobiana con un patrón característico, que aunque varía entre individuos o regiones geográficas, muestra cierta tendencia caracterizada por la presencia de un grupo de bacterias gramnegativas anaerobias consideradas como periodontopatógenas, entre las cuales se han identificado Porphyromonas gingivalis, Actinobacillus actinomycetemcomitans, Tannerella forshytia, Eikenella corrodens, Campylobacter rectus, Micromonas micros, Treponema denticola y Prevotella intermedia2.
Los productos de las bacterias periodontopatógenas, en especial el lipopolisacárido (LPS) reta al sistema inmune del hospedero, y genera una respuesta inmune inflamatoria tanto inespecífica como específica (Th1/Th2)3. En esta respuesta, el aumento de citoquinas pro-inflamatorias (IL 1 alfa, IL-6, TNF-alfa) es importante para activar el sistema inmune y el control de la infección. Sin embargo, estas mismas citoquinas pueden tener pleiotropismo por varias poblaciones celulares del periodonto (células epiteliales y dendríticas, fibroblastos, linfocitos y células polimorfonucleares), y originar así comportamientos celulares distintos. Esto logra afectar la función celular de estas células para que produzcan algún tipo de respuesta pro-inflamatoria que finalmente puede resultar en la resorción del tejido conectivo (incluyendo el hueso alveolar) y dar lugar a la enfermedad periodontal4.
Una población que tiene un papel importante en la respuesta inmune periodontal son los fibroblastos gingivales humanos (FGH)4. Los FGH son las células de origen mesenquimal predominantes en los tejidos conectivo gingival y periodontal; están comprometidos en el funcionamiento normal y anormal de los tejidos periodontales en su desarrollo, mantenimiento, reparación y defensa4. Los fibroblastos alteran sus funciones normales en respuesta a citoquinas pro-inflamatorias. En la lesión inicial gingival5 una de las características más importantes es la reducción del número de fibroblastos por campo. En teoría se desconoce la explicación a este fenómeno. Una hipótesis sugiere que los fibroblastos se pueden disminuir y/o eliminar por medio de la apoptosis («muerte celular programada»). La apoptosis es un fenómeno que forma parte del desarrollo embriológico y del ciclo celular de los organismos multicelulares, y su expresión se controla fisiológicamente o también se puede activar en respuesta a un estímulo patológico6. Las células pueden morir ante una amplia variedad de estímulos, pero en la apoptosis esta muerte ocurre de una manera regulada e individual, proceso que se diferencia notoriamente de la necrosis, donde se genera lisis celular, respuestas inflamatorias inespecíficas y degeneración tisular irreversible. Cuando una célula recibe una señal específica para empezar el proceso apoptótico se inicia una serie de cambios bioquímicos y morfológicos celulares. Específicamente, un grupo de proteínas conocidas como las caspasas se activan en los estadíos tempranos de la apoptosis. Estas proteínas se encargan de hacer un clivaje o corte específico de los sustratos celulares comprometidos en la función celular normal como algunas proteínas estructurales del citoesqueleto (cromatina) o ciertas proteínas nucleares (enzimas reparativas del ADN). Luego la célula empieza a reducir su tamaño normal y se empaqueta para formar los cuerpos apoptóticos, de tal manera que se facilite su depuración o eliminación a cargo de los macrófagos tisulares7.
Los estímulos pro-apoptóticos pueden ser de tipo intrínseco (regulados por la mitocondria) o extrínsecos (moléculas agonistas que actúan como ligandos de los «receptores de la muerte»)8. La vía extrínseca cobra importancia en los procesos inflamatorios debido a que la apoptosis se puede activar cuando la célula no está preparada para hacerlo. Los «receptores de la muerte» una vez urgidos por ligandos agonistas específicos, transmiten una cascada de señales bioquímicas que agilizan la vía de las caspasas en cuestión de segundos. Algunos receptores de la muerte pertenecen a la familia del factor de necrosis tumoral (TNF-R1) activados por los ligandos TRAIL (TNF-related apoptosis inducing ligand o ligando inductor de apoptosis relacionado con TNF), o el CD-95 (receptor Fas)9.
Por ende, los FGH interactúan con los productos bacterianos y podrían inducir apoptosis en respuesta a la activación de la vía extrínseca. El propósito de este estudio es determinar y cuantificar la apoptosis de los FGH en biopsias del periodonto de individuos sanos, así como de sujetos con gingivitis y con periodontitis crónica.
MÉTODOS
Se realizó un estudio clínico descriptivo de corte transversal en pacientes consecutivos de las clínicas odontológicas de la Escuela de Odontología de la Universidad del Valle, previa firma de un consentimiento informado que aprobó el Comité de Ética en Salud Humana de la Facultad de Salud de la Universidad del Valle. Se incluyeron pacientes con diagnóstico clínico de salud periodontal (S), gingivitis (G) y periodontitis crónica (PC) en tratamiento periodontal y programados para recibir procedimientos quirúrgicos dentro de su plan terapéutico. Se excluyeron pacientes con tratamiento periodontal previo en un período menor a 6 meses, fumadores, con alguna enfermedad sistémica incluyendo diabetes, enfermedades autoinmunes, síndromes de Chediak-Higashi, Papillon-Lefevre o retardo mental (trisomías). También se excluyeron los pacientes con terapia antibiótica y/o anti-inflamatoria en un período menor a un mes previo al estudio.
Se hizo un examen periodontal completo a cada uno de los sujetos incluidos. Se registraron en la base de datos los siguientes parámetros clínicos periodontales: índice de placa bacteriana, índice gingival (10,11), sangrado al sondeo periodontal, profundidad de surco/bolsa y el nivel clínico de inserción. Todas las mediciones se realizaron con una sonda periodontal milimetrada (UNC-15, Hu Friedy, EE.UU.) mediante presión manual leve. El diagnóstico periodontal se categorizó según los criterios de la Academia Americana de Periodontología (12) que aparecen en el Cuadro 1.
Obtención de las biopsias periodontales. A quienes ingresaron en este estudio, escogidos por conveniencia, se sometieron a distintos procedimientos quirúrgicos dentro de su plan de tratamiento periodontal (gingivectomía, cuñas mesiales o distales, alargamientos de corona clínica y eliminación quirúrgica de la bolsa periodontal), facilitando así la obtención de la biopsia escisional. El tejido periodontal retirado se aprovechó como muestra biológica para su posterior análisis patológico e inmunohistoquímico.
Procesamiento del tejido obtenido. Se obtuvieron las biopsias periodontales bajo anestesia local y se almacenaron inmediatamente en un recipiente de vidrio con formaldehído amortiguado al 10% para su fijación. Las biopsias se transportaron al laboratorio de tejidos del Departamento de Patología de la Universidad del Valle, donde se sumergieron en parafina para cortarlas sagitalmente con un micrótomo especial a un grosor de 5 µm. Los cortes se montan en portaobjetos previamente tratados con adhesivo tisular (Poly-A) y se dejan secar a temperatura ambiente durante 24 horas. Al siguiente día los tejidos se desparafinan en gradientes de xilol y se hidratan en alcoholes decrecientes, con un último paso en agua destilada y se sumergen en el medio de rescate. Después de un proceso previo de estandarización se escogió el rescate por medio de autoclave a 30 lbs de presión y 125ºC, durante 15 minutos en medio de rescate EDTA a pH 8.0 (13,14).
Inmunotinción. Una vez hecho el rescate las placas se enfrían con agua de la llave durante 30 min, y se sumergen en PBS amortiguado por 30 min más. Posteriormente se agrega peróxido de hidrógeno al 3% para el bloqueo de la enzima peroxidasa endógena, se hacen lavados en PBS amortiguado y se agregan 100 µl del anticuerpo primario diluidos en BSA (suero albúmina bovina: contiene suero de caballo, tritón y azida de sodio). Los anticuerpos primarios utilizados se presentan en el Cuadro 2; la incubación se realiza durante 24 horas bajo refrigeración. Luego se agregan los anticuerpos secundarios específicos para cada anticuerpo primario: para la caspasa 3 (IgG de conejo 1:600) o para la vimentina (IgG de ratón 1:600) durante 20 minutos. Se completa la técnica del sándwich con el conjugado de estreptavidina-biotina (Vectastain® ABC complex, Vector Labs, EE.UU.) y se revela con dia-minobenzidina (DAB substrate, Dako DIN). Por último, las placas se deshidratan y se contrastan con hematoxilina-eosina. Para la lectura se montan en un medio permanente con cubreobjetos (Permount, Fisher Scientific, EE.UU.)
En algunas placas se efectúa un procedimiento de doble tinción caspasa-vimentina con el propósito de diferenciar los fibroblastos gingivales apoptóticos en un mismo campo, mediante la técnica de complejos solubles de fosfatasa alcalina y anticuerpos de ratón anti-fosfatasa alcalina (naftol AS-MX fosfato, Levamisol) y rojo rápido (fast red). Por último las placas se fijan en un medio temporal sin cubreobjetos (fijación acuosa).
En definitiva, para cada paciente se hicieron las siguientes tinciones: hematoxilina-eosina (evaluación cualitativa del tipo de inflamación, cantidad de infiltrado, características morfológicas del epitelio, cantidad de colágeno y disposición de fibroblastos gingivales), inmunomarcación de caspasa-3 (evaluar cualitativa y cuantitativamente los fibroblastos y las células infiltradas) inmunomarcación de vimentina (evaluación cualitativa/cuantitativa de fibroblastos en cada uno de los tejidos) y doble tinción caspasa-vimentina (visualización específica de fibroblastos apoptóticos).
LECTURAS DE PLACA E INTERPRETACIÓN DE HALLAZGOS HISTOPATOLÓGICOS
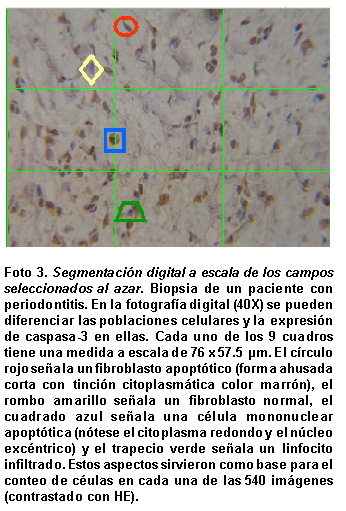
Digitalización, análisis y escalamiento de imágenes . Todas las placas se fotografiaron digitalmente (Canon Powershot A5, Canon, EEUU) a 4 megapixeles en el microscopio óptico de luz (OLYMPUS CH) a distintos aumentos (4X, 10X, 40X y 100X) para documentar todas las imágenes de las biopsias procesadas. Posteriormente se estandariza una toma de 3 segmentos de tejido conectivo escogidos al azar a un aumento de 40X para el análisis y recuento bajo un software especializado (SigmaScan Pro 5.0, SPSS Inc, EEUU). Los operadores de esta digitalización desconocen el objetivo del estudio y el diagnóstico periodontal de cada una de las biopsias. Mediante este software se estandarizaron las imágenes a 40X (1,600 x 1,200 píxeles) correspondientes a un campo visual de aproximadamente 230 µm x 172.5 µm. A su vez, para facilitar el recuento de células apoptóticas se aplicó a la imagen una cuadrícula digital que dividió el campo en 9 segmentos iguales, lo que dio origen a 9 campos por imagen de 76 µm x 57.5 µm (533.3 x 400 píxeles). De esta manera se obtuvieron 540 campos para analizar.
Análisis cualitativo y cuantitativo de los hallazgos histopatológicos. Además de los recuentos celulares bajo el software, las placas las interpretó histopatológicamente un médico patólogo. Este observador desconocía el diagnóstico periodontal clínico de las biopsias. Los siguientes parámetros se analizaron en cada una de las biopsias: tipo de inflamación (presencia de neutrófilos y/o la presencia de plasmocitos y linfocitos), densidad del infiltrado inflamatorio (cantidad de células inflamatorias en el tejido conectivo) hiperplasia epitelial (morfología del epitelio), vascularización (ausencia o angiogénesis), densidad en la expresión de vimentina y la relación del recuento total de fibroblastos/fibroblastos apoptóticos (porcentaje de fibroblastos apoptóticos/total de fibroblastos por biopsia).
Análisis estadístico. Se aplicó una estadística descriptiva a los datos obtenidos (promedio, error estándar de la media, frecuencias, etc.). Para las variables categóricas se aplicó la prueba del Chi2. La diferencia en las variables nominales (parámetros clínicos periodontales) entre los grupos se analizaron con la prueba de Kruskall-Wallis como prueba estadística no paramétrica. Para el análisis de la expresión de células apoptóticas vs. diagnóstico periodontal se aplicó la prueba ANOVA de dos vías. Además, se efectuó un análisis de regresión lineal entre el porcentaje de expresión de la apoptosis y la profundidad de la bolsa periodontal. En todas las pruebas estadísticas aplicadas se asumió una diferencia estadísticamente significante cuando p<0.05. Todos los datos se analizaron y graficaron con un software estadístico (GraphPad Prism versión 4.00 para Windows®, GraphPad Software, San Diego EEUU).
RESULTADOS
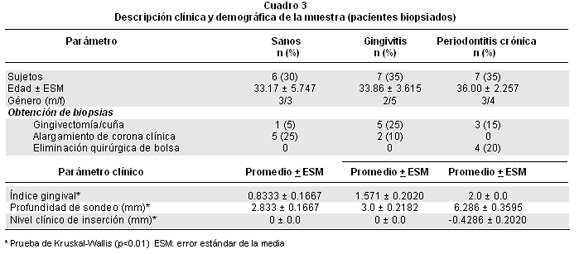
Descripción clínica y demográfica de la muestra (biopsias de los pacientes). El Cuadro 3 indica los datos clínicos y demográficos de los participantes incluidos en este estudio. Se estudiaron 20 personas (8 hombres y 12 mujeres) con distintos diagnósticos periodontales. A fin de obtener la biopsia periodontal se siguieron razones estéticas en los individuos sanos y con gingivitis, en contra del procedimiento usado para la mayoría de los pacientes con periodontitis crónica (eliminación quirúrgica de la bolsa o gingivectomía). Tanto el índice gingival, como la profundidad de sondeo y el nivel clínico de inserción fueron distintos entre los tres grupos (p<0.01). Los pacientes con periodontitis crónica presentaron mayor inflamación, más profundidad al sondeo y pérdida de inserción periodontal (-0.4±0.2), es decir hubo mayor recesión gingival en estos casos (explicado por el valor negativo del nivel clínico de inserción). No hubo ningún diagnóstico de periodontitis agresiva entre quienes se incluyeron en este estudio.
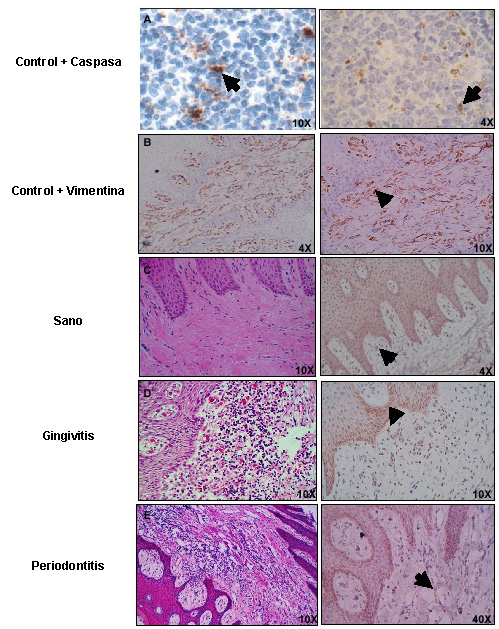
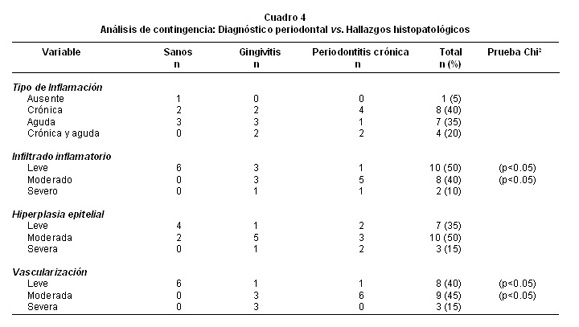
Análisis cualitativo de los hallazgos histopatológicos. En la Foto 1 se ilustran las biopsias más representativas de los sujetos estudiados. El Cuadro 4 ofrece el análisis de contingencia para el diagnóstico periodontal y los hallazgos histopatológicos. Sólo un individuo sano tuvo ausencia de inflamación histopatológica, en los demás la inflamación crónica fue la más común (40%), seguida de la aguda (35%) o la presencia de ambas (20%), sobre todo de tipo crónico con focos inflamatorios agudos. En cuanto a la cantidad de infiltrado inflamatorio los individuos sanos presentaron apenas infiltrados leves, lo que permitió establecer diferencias significativas entre los grupos (p<0.05, S vs. G/PC). La hiperplasia epitelial fue un hallazgo habitual en todas las biopsias estudiadas, y no hubo dife-rencias en su expresión de acuerdo con su frecuencia en cada uno de los grupos estudiados. Se encontró vascularización en todos los participantes, y su expresión fue diferente en la significancia entre los grupos (p<0.01, S vs. G/PC), pues era leve en los sujetos sanos y aumentó a moderada y severa en los casos de gingivitis y periodontitis. No hubo diferencias significativas entre el grupo gingivitis y periodontitis.
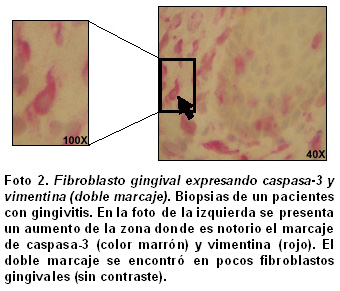
Análisis cuantitativo de los hallazgos histopatológicos. En la Foto 2 se presenta un ejemplo de la doble tinción caspasa-vimentina para diferenciar los fibroblastos gingivales apoptóticos en un mismo campo de un paciente con gingivitis. La marcación doble se pudo demostrar en pocos fibroblastos gingivales. Para detectar los fibroblastos apoptóticos y diferenciarlos de las otras poblaciones celulares se utilizaron criterios morfológicos y la expresión de caspasa-3 en ellas (formas ahusadas y cortas con tinción citoplasmática, citoplasma redondo y núcleos excéntricos, Foto 3).
El resultado de esta cuantificación aparece en la Gráfica 1 y 2 . La población celular total de fibroblastos tuvo un promedio de 430±67.6 en los pacientes sanos, y una disminución significativamente progresiva en gingivitis (270±37.1) y periodontitis crónica (206.5±69.8) (p<0.05, Gráfica 1 ). En cuanto a la población total de células del infiltrado inflamatorio se notó un aumento en gingivitis (191.8±50.1) y una disminución en periodontitis (109.3±21.7) sin haber significancia estadística. La expresión de fibroblastos apoptóticos por campo aumentó de acuerdo con la severidad de la enfermedad [28±16 en sanos (6.5%); 31±17 en gingivitis (11.5%) y 51±24 en periodontitis (24.8%), Gráfica 2, p<0.001]. Algo similar ocurrió para las células inflamatorias pero con porcentajes distintos de expresión [17±13 en sanos (23%); 28±19 en gingivitis (14.6%) y 47±35 en periodontitis (43.1%), Gráfica 2, p<0.05].
Con una prueba de regresión lineal se evaluaron las relaciones entre los porcentajes en que se expresan la apoptosis y la profundidad de la bolsa periodontal; se se encontró que para los fibroblastos apoptóticos marcados para caspasa-3 hubo una relación directamente proporcional baja sin significancia estadística (p>0.5, r²=0.02, Gráfica 3); para las células inflamatorias del infiltrado la relación fue directamente proporcional, baja pero significativa (p<0.05, r²=0.2018, Gráfica 4).
DISCUSIÓN
Para que los microorganismos colonicen el periodonto y alteren su función, deben poseer factores de virulencia capaces de sobrepasar las defensas del huésped e iniciar un proceso infeccioso. Muchos de estos factores son mecanismos que promueven la colonización y persistencia en la cavidad oral como las adhesinas, bacteriocinas, leucotoxinas, inhibidores quimiotácticos, proteínas inmunosupresoras, citotoxinas, colagenasas, agentes de reabsorción ósea, estimuladores de mediadores inflamatorios, factores inhibidores de la proliferación de fibroblastos e inhibidores de formación ósea (15). De acuerdo con los hallazgos obtenidos en este estudio la apoptosis se presenta en células epiteliales, en células del infiltrado inflamatorio y en FGH. Esto confirma los resultados de otros estudios que han evaluado la apoptosis en distintas poblaciones celulares (16-25).
En esta investigación un patólogo con experiencia calificó y cuantificó los hallazgos histopatológicos para cada una de las biopsias periodontales; esta información se contrastó con los recuentos en cantidad. En el Cuadro 4 se presenta el análisis de contingencia de los hallazgos histopatológicos vs el diagnóstico periodontal. La descripción histopatológica de la inflamación aguda incluye el reconocimiento de células de tipo neutrófilos.
En la inflamación crónica se identifican plasmocitos y linfocitos. También se pueden encontrar células que sugieren un estado inflamatorio crónico con focos agudos de inflamación. El resultado del análisis de contingencia arrojó diferencias sin significancia entre los tres diagnósticos, incluyendo el análisis para los grupos gingivitis y periodontitis (p>0.05). Solamente en una biopsia de un paciente hubo ausencia histológica de inflamación. En la mayoría la inflamación crónica fue la más común. Esto está de acuerdo con la literatura, donde se ha descrito que la enfermedad periodontal tiene características celulares y humorales (Th1 y Th2) (26). Lo controversial en enfermedad periodontal es si una tendencia hacia la inmunidad celular se sobrepone a una humoral, o viceversa. Es claro que tanto la respuesta inmune inespecífica como específica juegan un papel fundamental en la respuesta inmune periodontal ante el ataque bacteriano. Hubo una situación distinta al categorizar cualitativamente la cantidad de infiltrado inflamatorio donde se hubo diferencias significantes entre los grupos (p<0.05). Los individuos sanos presentaron un leve infiltrado inflamatorio, lo que permitió establecer diferencias significativas. Sin embargo, entre los grupos gingivitis y periodontitis no se vieron estas diferencias. Aun así, esta categorización afirma los resultados obtenidos entre los recuentos en cada una de las biopsias. En cuanto a hiperplasia epitelial las diferencias entre los grupos no tuvieron significancia (p=0.242), la hiperplasia epitelial fue un hallazgo en todas las biopsias del estudio. La hiperplasia epitelial es el resultado de la respuesta inflamatoria de las células epiteliales al ataque bacteriano y el epitelio de unión es uno de los tejidos con mayor capacidad de respuesta proinflamatoria (27). La hiperplasia leve en individuos sanos sugiere la actividad inmune del periodonto normal. En cuanto a la vascularización hubo diferencias significativas entre los grupos estudiados. La vascularización se acentuó en los pacientes con gingivitis y periodontitis como se ha descrito en lesiones periodontales extensas y se atribuye al factor de crecimiento vascular endotelial (VEGF) que puede ser producido por los FGH en respuesta a bacterias periodontopatógenas (28).
El análisis morfológico e histopatológico de las 20 biopsias permitió diferenciar claramente a nivel tisular las características descritas en estudios clásicos de la literatura periodontal (29-37). La disposición y densidad de fibroblastos se determinó por la marcación con el anticuerpo monoclonal vimentina (Foto 1). La vimentina es una proteína estructural de citoesqueleto (filamento intermedio tipo III), y cumple funciones relacionadas con crecimiento celular y/o mantenimiento tisular en células de origen mesenquimal exclusivamente (fibroblastos, miocitos, células gliales, etc.) (38). En la gran mayoría de las biopsias de individuos sanos fue notoria la disposición alargada y abundante de los fibroblastos gingivales en el tejido conectivo. También fue posible diferenciar la alteración de la morfología normal de los fibroblastos en las biopsias de los otros dos grupos, pues se vio que estaban menos ahusados y disminuidos en su población celular en comparación con las biopsias de individuos sanos.
La marcación para caspasa-3 se identificó en distintas poblaciones celulares incluyendo los fibroblastos. La doble tinción caspasa-3 y vimentina permitió diferenciar algunos fibroblastos apoptóticos en las biopsias estudiadas, pero no con la misma frecuencia en la marcación con caspasa-3 (Foto 2 ). Para compensar esta situación se hizo el recuento de las poblaciones celulares apoptóticas con la ayuda de un software especializado para diferenciar las poblaciones celulares (fibroblastos y células inflamatorias) y la expresión de caspasa-3 en ellas dentro del tejido conectivo. En cada campo (casi 4 mm2) se diferenciaron fibroblastos apoptóticos (forma ahusada corta con tinción citoplasmática color marrón), fibroblastos no apoptóticos (forma ahusada con tinción del núcleo por la HE), polimorfonucleares apoptóticos (citoplasma redondo, núcleos excéntricos lobulados con tinción color marrón) y células infiltradas no apoptóticas (citoplasma redondo con núcleo diferenciable por la HE). Estos aspectos sirvieron como base para el recuento de células en cada una de las 540 imágenes analizadas (Foto 3).
Cuantitativamente la expresión de caspasa-3 fue mayor en los grupos de biopsias con gingivitis y enfermedad periodontal (Gráfica 1). En la Gráfica 2 se muestra el recuento promedio por campo de fibroblastos apoptóticos y células inflamatorias apoptóticas en las biopsias de pacientes en los 3 grupos. En los fibroblastos apoptóticos la diferencia promedio por campo entre el grupo sano y gingivitis no fue significante (28±16 vs. 31±17) pero sí lo fue para el grupo periodontitis (51±24, p<0.001, prueba ANOVA de dos vías). Esto confirma los hallazgos de otros grupos que han descrito apoptosis en fibroblastos gingivales de pacientes con enfermedad periodontal (17,39). Este es el primer trabajo que cuantifica fibroblastos apoptóticos en tres grupos de diagnóstico periodontal distinto y pone en evidencia un aumento en la expresión de apoptosis en los fibroblastos gingivales en relación con el desarrollo de la etiopatogénesis de la enfermedad periodontal. Un estudio reciente40 evaluó la activación de caspasas en enfermedad periodontal, pues indicó que la activación de caspasa-3 estaba aumentada considerablemente en biopsias de 18 pacientes con enfermedad periodontal en comparación con tejidos sanos de 11 individuos sin enfermedad periodontal, siendo las células del epitelio oral, queratinocitos, células epiteliales basales y células del tejido conectivo subyacente las inmunorreactivas más comunes; sin embargo, no se evaluó de modo específico la apoptosis en fibroblastos gingivales.
En relación con el recuento de las células infiltradas apoptóticas, hubo diferencias significativas entre los grupos sanos, gingivitis y periodontitis (17±13, 28±19 y 47±35) respectivamente (p<0.001, prueba ANOVA de dos vías). Estos hallazgos confirman los de otros estudios (21,41). En un informe sobre enfermedad periodontal (42) se vio una apoptosis disminuida en neutrófilos. Una explicación a este hallazgo es la depuración de las células infiltradas como mecanismo de parada de la respuesta inmune innata, que aumenta con la cronicidad de la infección (43). La evidencia científica es contradictoria; muchas de las bacterias periodontopatógenas tienen factores de virulencia que retardan la apoptosis de las células infiltradas (44-49), o por el contrario sus productos bacterianos pueden inducirla (50-62).
En este estudio es de interés la expresión de fibroblastos apoptóticos/campo en individuos sanos y la falta de diferencias significativas en la expresión de los pacientes con gingivitis. Si bien la expresión de fibroblastos apoptóticos/campo en individuos sanos es similar a la de la gingivitis, la proporción de fibroblastos no apoptóticos en sujetos sanos por campo es mayor (6.5% vs. 11.5% respectivamente). Por tanto, se debe investigar más el umbral de diferencia entre normalidad de apoptosis como proceso fisiológico vs. la activación extrínseca de la apoptosis en distintas poblaciones celulares.
En la Gráfica 3 se presenta el análisis de regresión lineal para evaluar las relaciones entre la proporción de expresión de caspasa-3 en fibroblastos vs la profundidad de la bolsa, en un intento de calcular alguna correlación con la severidad de la enfermedad periodontal. Aunque esta correlación tuvo una tendencia directamente proporcional, no es significante (p>0.5, r²=0.02). Cuando se evalúa el mismo parámetro con la proporción de expresión de caspasa-3 en células infiltradas se encuentra una relación directamente proporcional significativa con baja correlación (p<0.05, r²=0.2018). Aunque la profundidad de la bolsa es el factor clínico más usado para medir el progreso de la enfermedad periodontal, junto con la pérdida de inserción clínica, no necesariamente se relaciona en forma directa con descenso en el número de fibroblastos (63). En cambio, la profundidad de la bolsa provee un muy buen estimativo y se correlaciona estrechamente con la concentración de mediadores inflamatorios y células infiltradas comprometidas en la patogénesis de la enfermedad periodontal (Gráfica 4) (64,65). El hallazgo de una correlación significativa entre la profundidad de la bolsa y el aumento en la apoptosis de células infiltradas podría ser tema de futuras investigaciones.
Esta investigación proporciona una evidencia científica con respecto a los FGH y su comportamiento como células inmunocompetentes que ocupan un lugar notorio dentro de la respuesta del hospedero ante la enfermedad periodontal. Como son las células más numerosas en el periodonto, su papel puede ser importante y valioso, y la apoptosis inducida por factores extrínsecos puede ser uno de los mecanismos que explique su disminución en número en el comienzo de la lesión gingival. Es de interés conocer aún más esta célula, profundizar en el entendimiento de su papel dentro de la etiopatogénesis de la enfermedad periodontal y su perspectiva terapéutica en el futuro.
Los resultados en esta investigación permiten concluir que tanto los FGH como las celulas inflamatorias presentan apoptosis manifiesta por la expresión de caspasa-3, y ésta se incrementa de modo significativo en los grupos de gingivitis y enfermedad periodontal; y en relación directa con la enfermedad periodontal y con la profundidad de bolsa, respectivamente.
AGRADECIMIENTOS
Los autores desean agradecer a la Vicerrectoría de Investigaciones y la Escuela de Odontología de la Universidad del Valle por la co-financiación del proyecto. A los doctores Adolfo Contreras y Oscar Gutiérrez por su asesoramiento. Al Centro de Estudios Cerebrales de la Facultad de Salud por la ayuda en la cuantificación celular.
REFERENCIAS
1. Loe H, Theilade E, Jensen SB. Experimental gingivitis in man. J Periodontol 1965; 36: 177-187.
2. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RLJr. Microbial complexes in subgingival plaque. J Clin Periodontol 1998; 25: 134-144.
3. Yamamoto M, Fujihashi K, Hiroi T, McGhee JR, Van Dyke TE, Kiyono H. Molecular and cellular mechanisms for periodontal diseases: role of Th1 and Th2 type cytokines in induction of mucosal inflammation. J Periodontal Res 1997; 32:115-119.
4. Bartold PM, Walsh LJ, Narayanan AS. Molecular and cell biology of the gingiva. Periodontol 2000 2000; 24: 28-55.
5. Payne WA, Page RC, Ogilvie AL, Hall WB. Histopathologic features of the initial and early stages of experimental gingivitis in man. J Periodontal Res 1975; 10: 51-64.
6. Perl M, Chung CS, Ayala A. Apoptosis. Crit Care Med 2005; 33 (Suppl 12): 526-529.
7. Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 2005; 37: 719-727.
8. Kam PC, Ferch NI. Apoptosis: mechanisms and clinical implications. Anaesthesia 2000; 55: 1081-1093.
9. Chaudhari BR, Murphy RF, Agrawal DK. Following the TRAIL to apoptosis. Immunol Res 2006; 35: 249-262.
10. Loe H. The Gingival Index, the Plaque Index and the Retention Index Systems. J Periodontol 1967; 38 (Suppl 6): 610-616.
11. Greene JC. The oral hygiene index-development and uses. J Periodontol 1967; 38 (Suppl): 625-637.
12. International Workshop for a Classification of Periodontal Diseases and Conditions. Papers. Oak Brook, Illinois, 1999. Ann Periodontol 1999; 4: i1-i112..
13. Imam SA, Young L, Chaiwun B, Taylor CR. Comparison of two microwave based antigen-retrieval solutions in unmasking epitopes in formalin-fixed tissue for immunostaining. Anticancer Res 1995; 15: 1153-1158..
14. Pileri SA, Roncador G, Ceccarelli C, Piccioli M, Briskomatis A, Sabattini E, et al. Antigen retrieval techniques in immunohistochemistry: comparison of different methods. J Pathol 1997; 183: 116-123
15. Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000 1999; 20: 136-167.
16. Sorkin BC, Niederman R. Short chain carboxylic acids decrease human gingival keratinocyte proliferation and increase apoptosis and necrosis. J Clin Periodontol 1998; 25: 311-315.
17. Tonetti MS, Cortellini D, Lang NP. In situ detection of apoptosis at sites of chronic bacterially induced inflammation in human gingiva. Infect Immun 1998; 66: 5190-5195.
18. Koulouri O, Lappin DF, Radvar M, Kinane DF. Cell division, synthetic capacity and apoptosis in periodontal lesions analysed by in situ hybridisation and immunohistochemistry. J Clin Periodontol 1999; 26: 552-559.
19. Wang PL, Shirasu S, Shinohara M, Daito M, Oido M, Kowashi Y, et al. Induction of apoptosis in human gingival fibroblasts by a Porphyromonas gingivalis protease preparation. Arch Oral Biol 1999; 44: 337-342.
20. Arakawa S, Nakajima T, Ishikura H, Ichinose S, Ishikawa I, Tsuchida N. Novel apoptosis-inducing activity in Bacteroides forsythus: a comparative study with three serotypes of Actinobacillus actinomycetemcomitans. Infect Immun 2000; 68: 4611-4615.
21. Gamonal J, Bascones A, Acevedo A, Blanco E, Silva A. Apoptosis in chronic adult periodontitis analyzed by in situ DNA breaks, electron microscopy, and immunohistochemistry. J Periodontol 2001; 72: 517-525.
22. Graves DT, Oskoui M, Volejnikova S, Naguib G, Cai S, Desta T, et al. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res 2001; 80: 1875-1879.
23. Alikhani M, Alikhani Z, He H, Liu R, Popek BI, Graves DT. Lipopolysaccharides indirectly stimulate apoptosis and global induction of apoptotic genes in fibroblasts. J Biol Chem 2003; 278: 52901-52908.
24. Gamonal J, Sanz M, O'Connor A, Acevedo A, Suárez I, Sanz A, et al. Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol 2003; 30: 616-623.
25. Alikhani M, Alikhani Z, Raptis M, Graves DT. TNF-alpha in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol 2004; 201: 341-348.
26. Ebersole JL. Humoral immune responses in gingival crevice fluid: local and systemic implications. Periodontol 2000 2003; 31: 135-166.
27. Ekuni D, Tomofuji T, Yamanaka R, Tachibana K, Yamamoto T, Watanabe T. Initial apical migration of junctional epithelium in rats following application of lipopolysaccharide and proteases. J Periodontol 2005; 76: 43-48.
28. Suthin K, Matsushita K, Machigashira M, Tatsuyama S, Imamura T, Torii M, et al. Enhanced expression of vascular endothelial growth factor by periodontal pathogens in gingival fibroblasts. J Periodontal Res 2003; 38: 90-96.
29. Payne WA, Page RC, Ogilvie AL, Hall WB. Histopathologic features of the initial and early stages of experimental gingivitis in man. J Periodontal Res 1975; 10: 51-64.
30. Page RC, Offenbacher S, Schroeder HE, Seymour GJ, Kornman KS. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontol 2000 1997; 14: 216-248.
31. Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000 1997; 14: 9-11.
32. Page RC, Simpson DM, Ammons WF, Schectman LR. Host response in chronic periodontal disease. J Periodontal Res 1972; 7: 283-296.
33. Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest 1976; 34: 235-249.
34. Page RC, Schroeder HE. Current status of the host response in chronic marginal periodontitis. J Periodontol 1981; 52: 477-491.
35. Page RC. Current understanding of the aetiology and progression of periodontal disease. Int Dent J 1986; 36: 153-161.
36. Page RC. The etiology and pathogenesis of periodontitis. Compend Contin Educ Dent 2002; 23 (5 Suppl): 11-14.
37. Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontology 2000 1997; 14: 33-53.
38. Wang N, Stamenovic D. Mechanics of vimentin intermediate filaments. J Muscle Res Cell Motil 2002; 23: 535-540.
39. Koulouri O, Lappin DF, Radvar M, Kinane DF. Cell division, synthetic capacity and apoptosis in periodontal lesions analysed by in situ hybridisation and immunohistochemistry. J Clin Periodontol 1999; 26: 552-559.
40. Bantel H, Beikler T, Flemmig TF, Schulze-Osthoff K. Caspase activation is involved in chronic periodontitis. FEBS Lett 2005; 579: 5559-5564.
41. Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Characterization of cellular infiltrate, detection of chemokine receptor CCR5 and interleukin-8 and RANTES chemokines in adult periodontitis. J Periodontal Res 2001; 36: 194-203.
42. Gamonal J, Sanz M, O'Connor A, Acevedo A, Suárez I, Sanz A, et al. Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol 2003; 30: 616-623.
43. Fadeel B, Kagan VE. Apoptosis and macrophage clearance of neutrophils: regulation by reactive oxygen species. Redox Rep 2003; 8: 143-150.
44. Yamamoto M, Kawabata K, Fujihashi K, McGhee JR, Van Dyke TE, Bamberg TV, et al. Absence of exogenous interleukin-4-induced apoptosis of gingival macrophages may contribute to chronic inflammation in periodontal diseases. Am J Pathol 1996; 148: 331-339.
45. Gamonal J, Sanz M, O'Connor A, Acevedo A, Suárez I, Sanz A, et al. Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol 2003; 30: 616-623.
46. Kurita-Ochiai T, Ochiai K, Suzuki N, Otsuka K, Fukushima K. Human gingival fibroblasts rescue butyric acid-induced T-cell apoptosis. Infect Immun 2002; 70: 2361-2367.
47. Muro M, Nakashima K, Tomioka J, Kato S, Nonaka K, Yoshida T, et al. Inhibitory effect of lipopolysaccharide on apoptotic cell death in macrophages infected with Actinobacillus actinomycetemcomitans. FEMS Microbiol Lett 1999; 175: 211-216.
48. Crawford JM, Wilton JM, Richardson P. Neutrophils die in the gingival crevice, periodontal pocket, and oral cavity by necrosis and not apoptosis. J Periodontol 2000; 71: 1121-1129.
49. Preshaw PM, Schifferle RE, Walters JD. Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodontal Res 1999; 34: 197-202.
50. Nalbant A, Zadeh HH. Actinobacillus actinomycetemcomitans induces apoptosis of T lymphocytes by the Fas and Fas ligand pathway. Oral Microbiol Immunol 2002; 17: 277-284.
51. Ohguchi M, Ishisaki A, Okahashi N, Koide M, Koseki T, Yamato K, et al. Actinobacillus actinomycetemcomitans toxin induces both cell cycle arrest in the G2/M phase and apoptosis. Infect Immun 1998; 66: 5980-5987.
52. Yamamoto S, Mogi M, Kinpara K, Ishihara Y, Ueda N, Amano K, et al. Anti-proliferative capsular-like polysaccharide antigen from Actinobacillus actinomycetemcomitans induces apoptotic cell death in mouse osteoblastic MC3T3-E1 cells. J Dent Res 1999; 78: 1230-1237.
53. Gemmell E, Prajaneh S, Grieco DA, Taylor JJ, Seymour GJ. Apoptosis in Porphyromonas gingivalis-specific T-cell lines. Oral Microbiol Immunol 1999; 14: 331-338.
54. Kato S, Muro M, Akifusa S, Hanada N, Semba I, Fujii T, et al. Evidence for apoptosis of murine macrophages by Actinobacillus actinomycetemcomitans infection. Infect Immun 1995; 63: 3914-3919.
55. Nalbant A, Zadeh HH. Evidence for apoptosis of the majority of T cells activated in vitro with Actinobacillus actinomycetemcomitans. Oral Microbiol Immunol 2000; 15: 290-298.
56. Kato S, Nakashima K, Inoue M, Tomioka J, Nonaka K, Nishihara T, et al. Human epithelial cell death caused by Actinobacillus actinomycetemcomitans infection. J Med Microbiol 2000; 49: 739-745.
57. Sawa T, Nishimura F, Ohyama H, Takahashi K, Takashiba S, Murayama Y. In vitro induction of activation-induced cell death in lymphocytes from chronic periodontal lesions by exogenous Fas ligand. Infect Immun 1999; 67: 1450-1454.
58. Nonaka K, Ishisaki A, Okahashi N, Koseki T, Kato S, Muro M, et al. Involvement of caspases in apoptotic cell death of murine macrophages infected with Actinobacillus actinomycetemcomitans. J Periodontal Res 2001; 36: 40-47.
59. Mangan DF, Taichman NS, Lally ET, Wahl SM. Lethal effects of Actinobacillus actinomycetemcomitans leukotoxin on human T lymphocytes. Infect Immun 1991; 59: 3267-3272.
60. Brozovic S, Sahoo R, Barve S, Shiba H, Uriarte S, Blumberg RS, et al. Porphyromonas gingivalis enhances FasL expression via up-regulation of NF{kappa}B-mediated gene transcription and induces apoptotic cell death in human gingival epithelial cells. Microbiology 2006; 152: 797-806.
61. Sorkin BC, Niederman R. Short chain carboxylic acids decrease human gingival keratinocyte proliferation and increase apoptosis and necrosis. J Clin Periodontol 1998; 25: 311-315.
62. Volejnikova S, Marks SCJr, Graves DT. Tumor necrosis factor modulates apoptosis of monocytes in areas of developmentally regulated bone remodeling. J Bone Miner Res 2002; 17: 991-997.
63. Page RC. The etiology and pathogenesis of periodontitis. Compend Contin Educ Dent 2002; 23 (5 Suppl): 11-14.
64. Paquette DW, Williams RC. Modulation of host inflammatory mediators as a treatment strategy for periodontal diseases. Periodontol 2000 2000; 24: 239-252.
65. Paquette DW. Pocket depth reduction as an outcome measure of inflammation and soft tissue changes in periodontitis trials. J Int Acad Períodontol 2005; 7 (4 Suppl): 147-156.