Efecto potencial del ejercicio físico y del consumo de micronutrientes durante la gestación en factores maternos y placentarios asociados con enfermedades crónicas no transmisibles (ECNT) del adulto*
Isabella Echeverry, MD, MSc1, Robinson Ramírez-Vélez, FT1, José Guillermo Ortega, BACT1, Mildrey Mosquera, MSc1, Julio César Mateus, MD, MEpi2, Ana Cecilia Aguilar de Plata, MSc2
* Este estudio fue financiado por el Instituto Colombiano para la Ciencia y Tecnología «Francisco José Caldas» (COLCIENCIAS), Código N° 110645921540.
1. Grupo de Nutrición, Facultad de Salud, Universidad del Valle, Cali. Colombia. e-mail: isabella_ech@hotmail.com robin640@hotmail.com guileort@yahoo.com mmildrey@univalle.edu.co caplata@yahoo.es
2. Grupo de la División Salud, Fundación FES Social, Cali, Colombia. e-mail: jcmateus@fundacionfes.org
Recibido para publicación abril 10, 2009 Aceptado para publicación septiembre 30, 2009
RESUMEN
Introducción: En la actualidad casi todos los esfuerzos para prevenir las enfermedades crónicas no transmisibles (ECNT) a nivel poblacional, se han centrado en promover comportamientos saludables como el ejercicio, la actividad física, el consumo de frutas y verduras, y el desestimular el consumo de tabaco y alcohol en la población adulta, pero los resultados han sido poco alentadores. En los últimos años, múltiples estudios han señalado la relación entre alteraciones del crecimiento fetal y el desarrollo de ECNT en la edad adulta. Más recientemente, se ha propuesto que factores maternos (función endotelial, estrés oxidativo y alteraciones en adipoquinas) y placentarios (disfunción mitocondrial) pueden ser mecanismos precursores de alteraciones metabólicas fetales y del desarrollo posterior de ECNT y que intervenciones como el ejercicio físico y la complementación con micronutrientes durante la gestación podrían regular dichos factores maternos y placentarios.
Objetivo: Realizar una revisión de la literatura para verificar el papel del ejercicio físico y los micronutrientes durante la gestación sobre factores maternos y placentarios relacionados con ECNT del adulto.
Metododología: Se utilizaron las siguientes bases de datos: Medline, Scielo, EMBASE, Science Direct, Cochrane Central Register of Controlled Trials y The Cochrane Libraryp Pregnancy, fetal development, oxidative stress, vascular endothelium, mitochondrial, adipokines, micronutrients, exercise.
Resultados: El estrés oxidativo, como mecanismo central de otros eventos fisiopatológicos (alteración en los niveles de adipoquinas, disfunción endotelial y mitocondrial), tiene un papel importante en la programación fetal de ECNT, factores como la complementación con micronutrientes y el ejercicio físico, durante la gestación, podrían modular este estado y contribuir posiblemente a la prevención temprana de ECNT.
Conclusión: Se debe establecer si los mecanismos moleculares y fisiológicos propuestos están relacionados con alteraciones metabólicas fetales y si la complementación durante la gestación con micronutrientes y/o el ejercicio físico los pueden regular.
Palabras clave: Embarazo; Programación fetal; Estrés oxidativo; Función endotelial; Mitocondria; Adipoquinas; Ejercicio; Micronutrientes.
Potential effect of physical exercise and consumption of micronutrients during pregnancy on the placental and maternal factors associated with chronic non-communicable diseases in adult
SUMMARY
Introduction: Currently, most efforts to prevent nontransmissible chronic diseases at population level have centered on promoting healthy behaviors like physical activity, consumption of fruits and vegetables, and discouraging from the consumption of tobacco and alcohol in the adult population, but the results have been less than hopeful. During recent years, a number of studies have indicated the relation between metabolic alterations and fetal growth with the development of nontransmissible chronic diseases in adult age. More recently, it has been proposed that maternal factors (endothelial function, oxidative stress, and alterations in adipokynes) and placental factors (mitochondrial dysfunction) are the precursory mechanisms of fetal metabolic alterations and of the later development of nontransmissible chronic diseases. Also, it has been suggested that possibly supplementation with micronutrients and physical exercise during gestation could regulate these maternal and placental factors.
Aim: To conduct a literature review to verify the role of physical exercise and micronutrients during pregnancy on placental and maternal factors related to nontransmissible chronic diseases in adults.
Methods: Medline, SciELO, Embase, Science Direct, Cochrane Central Register of Controlled Trials, and the Cochrane Library were used in the last 10 years (1998-2008). The following topics were reviewed: pregnancy, fetal development, oxidative stress, vascular endothelium, mitochondrial dysfunction, adipokines, micronutrients, and exercise.
Results: Oxidative stress, as the central pathophysiological event, such as changes in levels of adipokynes, mitochondrial and endothelial dysfunction, plays an important role in fetal programming of chronic diseases and factors such as micronutrient supplementation and physical exercise during pregnancy could modulate this state in a charity institution aiding in the early prevention of chronic diseases.
Conclusion: To clarify whether the proposed molecular and physiological mechanism items are related to metabolic abnormalities and fetal complementation with micronutrients during pregnancy and/or regular physical exercise.
Keywords: Pregnancy; Fetal development; Oxidative stress; Endothelial function; Mitochondria; Adipokynes; Micronutrients; Exercise.
La morbi-mortalidad generada por las enfermedades crónicas no transmisibles-ECNT (enfermedad coronaria, diabetes mellitus tipo 2, hipertensión arterial y síndrome metabólico) es el principal problema de salud pública en el mundo. En las últimas décadas, se ha evidenciado cada vez más que alteraciones subyacentes a la generación y progreso de las ECNT en el adulto tienen lugar en la vida fetal1. Esto sugiere que además del estilo de vida (dieta, tabaquismo, actividad física) y el medio factores externos tradicionalmente asociados con ECNT en la vida post-natal, también existen factores relacionados con la gestación, los cuales pueden producir cambios metabólicos fetales que elevarían la probabilidad de presentar ECNT en la edad adulta.
Durante los últimos años se ha ido planteando el enfoque denominado «aproximación de curso de vida para la epidemiología de enfermedades crónicas» que busca establecer las relaciones entre factores endógenos (genéticos, metabólicos, etc) y factores externos involucrados en el desarrollo y progreso de las ECNT2. Este enfoque tiene como uno de sus objetivos identificar los periodos críticos durante los cuales ocurren cambios fundamentales para la aparición de una enfermedad e identificar también los periodos sensibles, en los cuales dichos cambios se pueden modular a través de intervenciones. Asimismo, este enfoque pretende esclarecer la manera en que se relacionan los factores externos y los endógenos para producir una ECNT en el transcurso de la vida de una o varias generaciones y así contribuir a la búsqueda de soluciones en la salud.
En relación con lo anterior, el embarazo se ha identificado como un periodo crítico para el desarrollo de ECNT puesto que factores endógenos maternos y/o placentarios, y factores externos pueden inducir en el feto cambios estructurales y/o metabólicos irreversibles o poco modificables en la vida post-natal, a lo cual se le ha dado el nombre, en las últimas décadas, de programación fetal de enfermedades crónicas.
Estos cambios estructurales y metabólicos fetales pueden relacionarse con factores externos como la dieta, el consumo de adictivos3, la actividad física4 y otros aspectos socio-económicos durante la vida post-natal para generar ECNT en la edad adulta. Además, se postula que estos cambios podrían ser detectados desde etapas tempranas de la vida5. Entre los factores de origen endógeno, la disfunción endotelial6 y mitocondrial, el estrés oxidativo y las alteraciones en los niveles de adipoquinas pueden estar comprometidos en una posible programación temprana de ECNT.
Como el embarazo es un período crítico en el desarrollo del individuo durante el cual hay cambios rápidos en la organización de sistemas y sub-sistemas biológicos, diversos factores pueden modificar estos cambios de manera favorable o no. En consecuencia, se postula que las intervenciones encaminadas a mejorar la disfunción endotelial y mitocondrial, el estrés oxidativo y las alteraciones en los niveles de adipoquinas pueden contribuir a prevenir la incidencia y progreso de las ECNT.
Por todo lo anterior, el objetivo de la presente revisión de literatura es discutir el papel del ejercicio físico y del consumo de micronutrientes sobre factores maternos y placentarios relacionados con ECNT del adulto.
MATERIAL Y MÉTODOS
La búsqueda se realizó entre julio y diciembre de 2008, y los artículos se identificaron mediante una búsqueda en la literatura de las bases de datos Medline, Scielo, EMBASE, Science Direct, Cochrane Central Register of Controlled Trials y The Cochrane Library, nco las palabras clave pregnancy, fetal development, oxidative stress, vascular endothelium, mitochondrial, adipokines, micronutrients, exercise. Los criterios de inclusión de los artículos fueron:
1. Artículos publicados entre 1998 hasta 2008, en inglés, portugués o castellano.
2. Artículos en humanos y modelos animales.
3. Artículos sobre resultados de trabajos experimentales.
4. Artículos con gestantes como población de estudio y con evaluación de los efectos de este estado sobre la función endotelial, el estrés oxidativo, la función mitrocondrial y/o los niveles de adipoquinas. O en los cuales se asociaran estas variables con el desarrollo de entidades propias de la gestación (preeclampsia, diabetes gestacional, etc) y/o con los resultados del embarazo (peso al nacer, tipo de parto, crecimiento o desarrollo fetal, etc.).
5. Artículos con otros tipos de población pero con evaluación del efecto de la suplementación con micronutrientes o el ejercicio físico sobre la función endotelial, el estrés oxidativo, la función mitrocondrial y/o los niveles de adipoquinas.
La búsqueda inicial con las palabras claves mencionadas arrojo 3,313 referencias, que disminuyeron a 2,488 de acuerdo con la fecha de publicación seleccionada. De estos 1,178 trabajos se hicieron en humanos o en animales y de los cuales 1,085 estaban publicados en Inglés, portugués, o castellano.
La segunda etapa del proceso consistió en la revisión de 1,085 resúmenes de los cuales 61 trabajos correspondían a trabajos experimentales y se utilizaron en la presente revisión.
RESULTADOS
Fundamentos de la hipótesis de la programación fetal de ECNT. En las últimas décadas se ha comprobado que existe asociación entre alteraciones en el crecimiento fetal y una mayor susceptibilidad a sufrir enfermedad crónica en la edad adulta, sugiriendo posible programación de ECNT en la vida fetal o neonatal temprana.
El término «programación» lo introdujo en la literatura científica en la década de 1970 Dörner de la Universidad de Humboldt-Alemania7 quien observó que los cambios en las concentraciones de hormonas, metabolitos y neurotransmisores durante periodos críticos del desarrollo, podían «pre-programar» el desarrollo cerebral, y producir alteraciones funcionales, enfermedades y síndromes metabólicos en el adulto. Sin embargo, la hipótesis de programación fetal de ECNT se aceptó en el ambiente científico durante la década siguiente con base en estudios epidemiológicos retrospectivos de grandes cohortes de adultos en Europa. Gracias a estos estudios, investigadores como Barker et al.8 demostraron la relación entre bajo peso y talla al nacer y aumento en el riesgo de sufrir enfermedades en el adulto como infarto agudo del miocardio y diabetes mellitus tipo 2. Otros estudios epidemiológicos en países de diversos lugares como Europa, Asia y América han confirmado esta relación y trabajos experimentales sobre todo en animales tratan de esclarecer sus mecanismos9.
Los modelos animales han mostrado que la restricción de nutrientes durante la gestación afecta de manera irreversible la estructura, fisiología y metabolismo de ciertos órganos «y programan» enfermedades en etapas futuras de la vida10.
Esta hipótesis de programación fetal se ha criticado por el tiempo que separa la causa y el efecto, durante el cual numerosas condiciones como nivel socio-económico, dieta y actividad física pueden determinar la ocurrencia de una ECNT. Es importante entonces, esclarecer mecanismos subyacentes a esta programación para entender mejor las asociaciones encontradas entre alteraciones del desarrollo fetal y ECNT en el adulto.
Factores fisiopatológicos posiblemente asociados con la programación fetal
Estrés oxidativo. Para explicar la programación fetal, una de las hipótesis es la programación que se debe a un estado de estrés oxidativo (EO)11. El EO se da cuando hay aumento en las concentraciones celulares de radicales libres de oxígeno (RLO) por un desequilibrio entre su producción y su eliminación por los sistemas antioxidantes biológicos. En condiciones fisiológicas, estos radicales actúan como moléculas de señalización que regulan la actividad de enzimas y la expresión de genes sensibles al estado de oxido-reducción de la célula. Sin embargo, cuando se encuentran en concentraciones elevadas pueden oxidar proteínas, lípidos y ácidos nucleicos con la consecuente alteración de la estructura y la función celulares.
El EO se halla comprometido en la etiología de diversas condiciones, como cáncer, diabetes y enfermedades cardiovasculares. Tambien se asocia con desnutrición o sobrealimentación y entidades del embarazo relacionadas con alteraciones del desarrollo fetal y prematurez, como preeclampsia, diabetes gestacional e infecciones. Por tanto, se considera el EO como uno de los posibles mecanismos subyacentes a la programación in utero de ECNT. En este aspecto se han hecho algunos estudios. Por ejemplo, Gupta et al.12 observaron aumento de marcadores de EO en neonatos que habían sufrido desnutrición intrauterina. Otros investigadores (Longini et al.13) encontraron aumento de estos marcadores en el líquido amniótico de gestaciones con retardo en el crecimiento intrauterino (RCIU). Además el estado de EO de la madre se ha correlacionado con el del feto. En modelos animales, se ha visto que la restricción dietaria, durante la gestación, aumenta la producción de RLO en las crías14.
De otro lado, el buen funcionamiento placentario es importante para el desarrollo fetal correcto y juega un papel básico al mediar los efectos del ambiente materno en el feto. Posiblemente, varias de las influencias nutricionales y endocrinas que después predisponen el feto a ECNT son mediadas por la placenta15, y factores endógenos como la placentación anormal (principal causa de RCIU) están comprometidos en la generación de EO. Lo anterior se da como consecuencia de la invasión uterina anormal del trofoblasto que disminuye la adecuada formación de los vasos sanguíneos y genera por último hipoxia placentaria y EO.
Según esta hipótesis, se considera que puede haber programación si ocurre una agresión oxidativa en periodos críticos del desarrollo, aun sin alteración del desarrollo fetal. Es decir, que neonatos con peso normal al nacimiento pueden sufrir programación de ECNT a pesar de no tener alteraciones antropométricas.
Como ciertos factores externos pueden generar EO en fetos y que también se ha encontrado un aumento de RLO en adultos con ECNT, se puede llegar a considerar que es uno de los mecanismos endógenos subyacentes a la generación de alteraciones fetales durante la gestación, al producir daños celulares progresivos que coadyuvan al desarrollo de ECNT en la vida adulta.
El EO, en la pared vascular, favorece el desarrollo de disfunción endotelial a través de diferentes mecanismos, como la activación de factores de transcripción proinflamatorios, por ejemplo el factor nuclear kappa B (NFkB) y la proteína activadora (AP-1)16, y la disminución en la biodisponibilidad del óxido nítrico, captado por el radical anión superóxido para formar otro RLO, como es el peroxinitrito. La disminución en los niveles del NO es uno de los mecanismos centrales en el desarrollo de disfunción endotelial. Además, el EO conduce a la oxidación de macromoléculas como las LDL oxidadas (LDLox) que generan toxicidad celular, inflamación, apoptosis y disfunción endotelial. También, los F2-isoprostanos, productos de la peroxidación lipídica, junto con la vasoconstricción y con el aumento de la agregación plaquetaria, se han asociado con la disfunción endotelial.
Disfunción endotelial. El endotelio es la monocapa de células que recubre el interior de los vasos sanguíneos, y por su función central en la regulación de la homeostasis vascular, juega un papel importante en la fisiopatología de las enfermedades crónicas cardiovasculares.
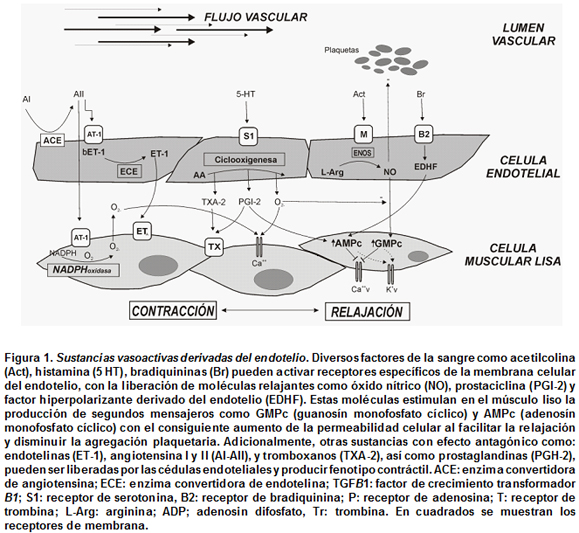
El endotelio provee una superficie anti-inflamatoria, anticoagulante, antiproliferativa y antitrombótica en estados fisiológicos. El principal mediador de la homeostasis vascular es el óxido nítrico (NO), sustancia implicada en la vasodilatación mediada por el endotelio17 y producido a partir de la L-arginina por la óxido nítrico sintasa endotelial (eNOS). La disminución en la biodisponibilidad del NO genera alteración de las funciones fisiológicas endoteliales y causa disfunción endotelial.
La eNOS es activada por el estrés de fricción del flujo sanguíneo en la pared vascular y por sustancias como acetilcolina, bradiquinina y serotonina. Está compuesta por dos proteínas globulares que al estar acopladas permiten la síntesis de NO y necesita para su actividad cofactores como tetrahidrobiopterina (BH4), NADPH, y de la unión de calmodulina asociada con calcio. Si se encuentra desacoplada puede producir anión superóxido (O2¯) en lugar de NO y ser fuente de estrés oxidativo18 (Figura 1).
Durante el embarazo factores como la desnutrición intrauterina, el cigarrillo y enfermedades como la preeclampsia, la hipertensión inducida por el embarazo y la diabetes gestacional19,20 se han asociado con disminución en la síntesis de NO y disfunción endotelial. Los estudios en ratas demostraron que la desnutrición intrauterina origina aumento de los valores de tensión arterial en las crías y disminución en la expresión y en la actividad de la eNOS en la aorta21. Andersen et al.22 encontraron que en células endoteliales de cordón umbilical, provenientes de recién nacidos, hijos de madres fumadoras, había disminución tanto en la actividad como en la concentración de la eNOS. Además, en las células endoteliales de cordón umbilical provenientes de embarazos complicados con preeclampsia, se ha disminuido la expresión y cantidad de la eNOS al comparar con embarazos normales23. También, en adolescentes con antecedente de bajo peso al nacer se observó la disminución de la vasodilatación mediada por el endotelio24, medida confiable de la función endotelial.
También, en el adulto, se vio una fuerte asociación entre disfunción endotelial y factores de riesgo aterogénicos. Por ejemplo, las lipoproteínas de baja densidad oxidadas (LDLox) disminuyen la expresión de eNOS y aumentan la producción del anión superóxido que favorece estados de EO. Igualmente, entidades como arteriosclerosis, síndromes isquémicos agudos y diabetes mellitus se relacionan con disfunción endotelial25.
Ésta, en consecuencia, puede mediar la asociación entre sucesos ocurridos durante la gestación y ECNT en el adulto. Sin embargo, todavía faltan estudios que evalúen en neonatos la función endotelial y su asociación con alteraciones del desarrollo fetal y el riesgo de desarrollar posteriormente una ECNT.
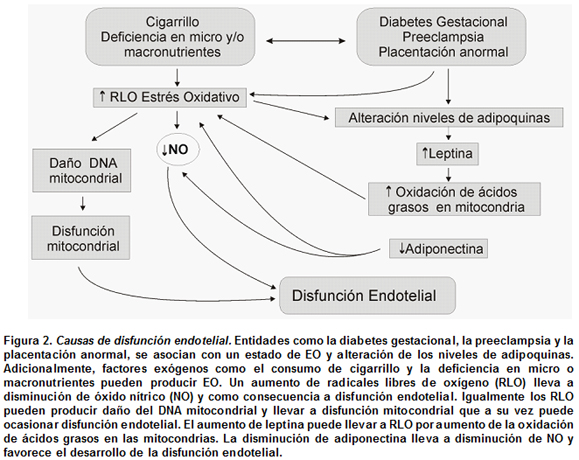
Disfunción mitocondrial. El EO también tiene que ver con otro estado fisiopatológico, la disfunción mitocondrial. La mitocondria es la organela celular encargada de producir más de 90% de la energía celular, en forma de ATP, por medio de la fosforilación oxidativa. Además, es la principal fuente de RLO, que son rápidamente degradados por enzimas antioxidantes como la superóxido dismutasa y la glutatión peroxidasa. La mitocondria posee su propio DNA, el DNA mitocondrial (mtDNA), que es mucho más vulnerable al daño que el DNA nuclear, pues no está protegido por histonas y no posee los mecanismos de reparación que se encuentran en el núcleo. Como mecanismo de generación de disfunción mitocondrial, se ha planteado que los estados de hiperglicemia o de aumento en los niveles de ácidos grasos circulantes, elevan la producción de radicales libres, disminuyen la formación de nuevas mitocondrias (biogénesis mitocondrial), y generan finalmente disfunción mitocondrial. Estos radicales pueden producir también daños directos en el mtDNA, en otras macromoléculas mitocondriales y alterar la función de la organela. El daño oxidativo mitocondrial podría aumentar con el paso de los años y desencadenar en ECNT. En modelos animales con desnutrición intrauterina se ha visto disminución del mtDNA en hígado, páncreas, músculo, y disminución de la expresión de genes mitocondriales26. En los seres humanos se adjunta correlación entre las concentraciones de mtDNA en los leucocitos de sangre periférica materna de cordón umbilical y el peso al nacer. Adicionalmente, en el adulto, condiciones como resistencia a la insulina y diabetes mellitus tipo 2 se unen a la disfunción mitocondrial27. Asimismo se notó que las células ß pancreáticas con menor cantidad de mtDNA liberan menos insulina28 y que hay menos mtDNA en el músculo de personas diabéticas. También, se ha visto correlación inversa entre la cantidad de mtDNA y los componentes del síndrome metabólico como hipertensión arterial, glucosa elevada en ayunas y circunferencia cintura-cadera29. Se propone que la disminución en la biogénesis mitocondrial conduce a la acumulación intracelular de lípidos, debido a la disminución en la fosforilación oxidativa, lo cual altera la señal a la insulina y se origina resistencia a esta hormona. Igualmente, las personas con alteraciones mitocondriales tienen complicaciones vasculares a temprana edad a pesar de no presentar factores de riesgo cardiovasculares conocidos; esto sugiere que la disfunción mitocondrial puede mediar el comienzo de la enfermedad cardiovascular al propiciar también el desarrollo de la disfunción endotelial30 (Figura 2).
Se propone que, la disfunción mitocondrial puede ser otro de los factores que relacionan la desnutrición fetal con alteraciones vasculares y del metabolismo de la glucosa en la vida adulta. Sin embargo, se requieren más estudios particularmente en seres humanos, para entender el detalle de los mecanismos de la disfunción mitocondrial y su papel en la programación del síndrome de resistencia a la insulina en el adulto o de otras ECNT.
Niveles de adipoquinas. Así como la placentación anormal ha sido asociada con la generación de estados de EO, también se ha asociado con alteraciones en la concentración de leptina y adiponectina placentarias. En la vida postnatal estas hormonas las secretan sobre todo el tejido adiposo y participan en la regulación del metabolismo, de la función cardiovascular y de la homeostasis energética entre otros papaeles. En el adulto, la leptina regula el consumo y el gasto energético a largo plazo, mientras que la adiponectina tiene propiedades anti-inflamatorias y eleva la sensibilidad a la insulina31.
Durante la gestación, la leptina «también conocida como la hormona de la saciedad,»producida por el tejido adiposo materno y la placenta, aumenta de modo progresivo hasta duplicarse alrededor de la semana 14 y permanece alta hasta el parto. Este aumento no altera el consumo energético materno, por lo que se considera el embarazo un estado de resistencia hipotalámica a la leptina. Además, en el feto, la leptina es un factor de crecimiento que aparece desde la etapa embrionaria y sube abruptamente entre las semanas 32-34 para coincidir con la expansión del tejido adiposo fetal.
Se ha observado que la hipoxia placentaria produce hiperleptinemia materna, lo que puede inducir también EO en las células endoteliales32 con la respectiva disminución de la biodisponibilidad del NO y finalmente la disfunción endotelial. El EO mediado por leptina parece que se relaciona con el alza en la oxidación de ácidos grasos en la mitocondria lo cual incrementaría la producción de RLO33 (Figura 2).
Otra adipokina relacionada y producida por el tejido adiposo materno y la placenta, es la adiponectina que se incrementa de modo gradual hasta la semana 14 y disminuye después del parto a un nivel similar al encontrado antes del embarazo.
Se ha observado que el EO y la hipoxia disminuyen la expresión de esta hormona en el adipocito, pero se desconoce su efecto en la placenta. La hipoadiponectinemia se asocia también con disfunción endotelial34 pues la adiponectina estimula la producción de NO.
Los niveles anormales de leptina y adiponectina se han unido a complicaciones durante el embarazo. En la preeclampsia y la diabetes gestacional se ha visto un aumento en los niveles de leptina y una disminución en los niveles de adiponectina. Tambien un aumento en las concentraciones de leptina en fetos y en las placentas de madres diabéticas y una disminución en los niveles de adiponectina en sus hijos al nacimiento. En el neonato los niveles de leptina y adiponectina se correlacionan de manera positiva con la cantidad de tejido adiposo. En neonatos con retardo en el crecimiento intrauterino (RCIU) los niveles de estas dos adipoquinas están disminuidos35. Sin embargo, al año de edad estos niños presentan niveles altos de leptina al compararlos con niños de peso adecuado al nacer. En adultos con antecedente de bajo peso al nacer ha habido niveles altos de esta hormona al compararlos con sujetos de igual índice de masa corporal.
En ratas sometidas a desnutrición, in utero, el aumento prematuro de leptina en la etapa postnatal temprana se asocia con obesidad en la adultez36. Además, el peso al nacer en los valores extremos bajo y alto se relaciona con un mayor porcentaje de grasa en etapas posteriores de la vida37. Posiblemente el nivel de adipoquinas en etapas tempranas del desarrollo juega un papel importante en la programación de la composición corporal del individuo. Asimismo, la alteración en los niveles de adipoquinas se une con varias enfermedades del adulto. Por ejemplo se ha observado hiperleptinemia en obesidad, síndrome metabólico, enfermedad cardiovascular y diabetes mellitus tipo 238.
Los anteriores hallazgos indican que las adipoquinas desempeñan papel importante en el desarrollo fetal y que posiblemente pueden participar, con los otros factores mencionados antes, para mediar una programación temprana de alteraciones como la obesidad y el síndrome metabólico. Sin embargo, se requieren aún más estudios que comprueben esta hipótesis.
Cambios en el estilo de vida: intervenciones con ejercicio físico y dieta. Varias investigaciones en modelos animales y en seres humanos, han demostrado que el ejercicio físico y la complementación con micronutrientes pueden modular diversos factores fisiopatológicos, entre ellos el EO, la función endotelial y mitocondrial, y los niveles de adipoquinas. No se sabe con certeza si el empleo de estas intervenciones durante la gestación podrían tener efectos benéficos tanto para la madre como para el feto, con un impacto favorable sobre una posible programación temprana de ECNT.
Ejercicio físico. El ejercicio físico (EF) provoca cambios fisiológicos que dependen tanto del tipo, la intensidad y la duración del esfuerzo, así como del entrenamiento físico, edad, género y estado nutricional del individuo en un momento dado.
En las embarazadas generalmente se observa que disminuyen su actividad física y que restringen el EF. Las actitudes relativas al EF durante el embarazo han sido moldeadas más por influencias culturales que por evidencias científicas. Con el avance del conocimiento, han surgido importantes preguntas sobre la relación riesgo/beneficio del ejercicio durante el embarazo, lo cual hace imprescindibles los consensos y recomendaciones para orientar políticas de salud pública.
Actualmente se tiene la certeza que la disminución tanto del oxígeno fetal como de la disponibilidad de carbohidratos durante el ejercicio, se acompaña de adaptaciones fisiológicas como el aumento de la extracción de oxígeno, la redistribución sanguínea intra-uterina y la hemoconcentración. Se sabe también, que el ejercicio regular produce múltiples beneficios al facilitar el trabajo de parto con disminución de su duración y de las complicaciones obstétricas39. Otros beneficios para la salud se relacionan con una menor probabilidad de padecer afecciones cardiovasculares y ciertas enfermedades durante el embarazo40, debido entre otros mecanismos a que el ejercicio incrementa el flujo sanguíneo, que a la vez eleva la tensión de roce o fricción de la sangre en el endotelio. Este proceso, al estimular la producción del NO41, favorece finalmente la vasodilatación. Estas conclusiones resultan de relacionar el ejercicio con el aumento en el diámetro de la arteria coronaria en estudios con animales, hallazgos en los cuales además, se ha visto que hay proporcionalidad con la intensidad del ejercicio. Otros hallazgos en la misma dirección, en sujetos físicamente activos al compararlos con sedentarios, son los niveles altos de metabolitos del NO y del cGMP (segundo mensajero del NO).
Otro beneficio del EF aeróbico en humanos, es el aumento de mitocondrias entre 50% a 100% en un periodo de 6 semanas42, concomitante con el incremento de la expresión del RNA mensajero (mRNA) de proteínas como: AMP kinasa (AMPK), citocromo c (proteína de la cadena transportadora de electrones), factores de transcripción como el factor A de transcripción mitocondrial (Tfam, el cual controla la expresión del DNA mitocondrial (mtDNA), factores nucleares de la respiración mitocondrial como c-jun y factor-1 (NRF-1), y el mtDNA que codifica proteínas de la cadena respiratoria. La actividad contráctil que implica el ejercicio físico es un estímulo que inicia una serie de hechos fisiológicos y bioquímicos que llevan a la biogénesis mitocondrial. Asimismo, se presentan cambios en el fenotipo celular, resultado de la adaptación metabólica inducida por el ejercicio físico, aunque su efecto (dependiente de la magnitud, intensidad y duración del EF) son todavía temas sin explicar.
Además se sabe que el EF regular de moderada intensidad, y comienzo precoz (primeras 20 semanas) en el embarazo, durante la fase hiperplásica del crecimiento placentario, aumenta la perfusión sanguínea placentaria y su capacidad funcional. De este modo, el EF puede prevenir la placentación anormal que es una de las causas subyacentes de la preeclampsia43 y que se da como consecuencia de una inapropiada invasión trofoblástica de las arterias espirales del útero al inicio del embarazo, que lleva a descenso de la perfusión celular e hipoxia placentaria. Como ya se dijo, el EO resultante causa disfunción endotelial sistémica que puede desencadenar hipertensión arterial y preeclampsia, pero también inducir el desarrollo de diabetes gestacional. Como el EF, renueva la perfusión placentaria, mejora el aporte de nutrientes necesarios para el desarrollo fetal, y de manera sustancial, disminuye el riesgo de desarrollar preeclampsia44.
En otros estudios, Covas et al. observaron que el ejercicio disminuye el EO mediante el aumento de enzimas antioxidantes como la superóxido dismutasa (SOD) y la glutation peroxidasa (GPX) en músculo esquelético, hígado, corazón, y sangre45. También se conoce la elevación en los niveles de la isoforma de la adiponectina cuya acción antiinflamatoria inhibe la proliferación celular y la producción de los RLO. Además, este desenlace aumenta durante el ejercicio, por la inhibición del factor de necrosis tumoral alfa (TNF-α), inhibidor de la adiponectina46.
Todos estos efectos unidos al incremento de la perfusión sanguínea placentaria, disminuyen el riesgo de disfunción endotelial sistémica y reducen así la probabilidad de preeclampsia y diabetes gestacional durante el embarazo.
La evidencia expuesta sugiere entonces que el EF en embarazadas puede ser un factor modulador de los mecanismos endógenos comprometidos en la generación de alteraciones metabólicas implicadas en la programación de ECNT. Se necesita más investigación para tener conclusiones definitivas.
Complementación con micronutrientes. El embarazo es un periodo de incremento en las demandas metabólicas, con cambios en la fisiología de la gestante y en las exigencias del feto que se forma. Los micronutrientes participan en todas las fases del progreso fetal, además son parte estructural de enzimas y organelas celulares. El desequilibrio de micronutrientes durante la gestación puede ocasionar alteraciones metabólicas tanto en la madre como en el feto, como consecuencia de cambios en enzimas, de factores de transcripción y en las vías de los procesos de señalización celular que regulan el desarrollo, y que finalmente pueden modificar el metabolismo in utero.
Algunos estudios en modelos animales demuestran que deficiencias nutricionales in utero programan diversas condiciones en la vida postnatal de las crías desnutridas. Se ha observado, que la restricción de minerales o vitaminas durante la gestación, produce, en las crías, aumento significativo en el porcentaje de tejido graso visceral, disminución del tejido magro y aumento de marcadores de EO en el plasma47. Estos cambios se unen con resistencia a la insulina y perfil de adipoquinas proaterogénicas durante la etapa adulta como hiperleptinemia e hipoadiponectinemia48.
Adicionalmente, a nivel poblacional y según la Organización Mundial de la Salud alrededor de 2000 millones de personas presentan deficiencia de micronutrientes con consecuencias socioeconómicas importantes49. En países como Estados Unidos gran proporción de mujeres en edad reproductiva consumen dietas que les proporcionan menos de las recomendaciones de micronutrientes como hierro, zinc, vitamina A y ácido fólico.
Se ha visto que la deficiencia de micronutrientes durante el embarazo en algunas poblaciones de Nepal, India, y Gambia es causa importante de bajo peso y talla al nacer50. Sin embargo, varios estudios sobre tales aspectos, donde se complementan esos elementosen las embarazadas, muestran resultados inconsistentes y el peso no parece un buen indicador para medir el efecto de la complementación, sobre todo en deficiencias moderadas.
En consecuencia, se requieren más estudios que evalúen el efecto de la complementación con micronutrientes durante la gestación sobre factores biológicos comprometidos en una posible programación fetal de ECNT.
Zinc, magnesio, calcio, vitamina A y niacina, previenen la disfunción endotelial, el EO y la alteración en los niveles de adipoquinas.Los estudios de la deficiencia de zinc en humanos la han asociado entre otras alteraciones con disminución de leptina plasmática51. En ratas hay disminución significativa de la expresión del gen o de leptina en tejido adiposo después de inducción de deficiencia marginal52. Esto permite suponer que la concentración de esta hormona se puede regular por los niveles de zinc. Su deficiencia también se encuentra unida a estados de EO, que se demuestra por el daño en moléculas como ADN, lípidos y proteínas53. Aún se desconoce si durante el período gestacional los efectos ocasionados por la deficiencia de zinc se mantienen y cuáles son sus implicaciones sobre la programación fetal.
El magnesio es un micronutriente esencial, cofactor de numerosas enzimas que participan en la defensa contra el EO. En ratas, las deficiencias de este mineral durante la gestación, han mostrado que las crías presentan mayor contenido de tejido adiposo visceral, resistencia a la insulina, intolerancia a la glucosa, disminución de leptina y aumento del TNF-α en la circulación54.
En cuanto al calcio, se ha visto que inhibe la lipogénesis, estimula la lipólisis y disminuye la adipogénesis. En modelos murinos la ingesta de calcio reduce la producción de RLO estimulado por dietas hipercalóricas55. Esta reducción del EO se asocia con el incremento en la expresión de factores antiinflamatorios como adiponectina e IL-15 y la diminución de citoquinas proinflamatorias como TNF-α e IL-6 en el tejido adiposo visceral56.
Las vitaminas como la niacina que forma parte de coenzimas esenciales en el metabolismo, participan en la defensa antioxidativa. In vitro, la escasez de esta vitamina se relaciona con el daño oxidativo de proteínas y ADN. En cultivo de keratinocitos se ha encontrado que la carencia de niacina aumenta la generación de RLO y la expresión de NADPH oxidasa, enzima generadora de RLO57. Adicionalmente, en los seres humanos, si hay suplementos de esta vitamina, aumentan en el suero entre 40% y 60% los niveles de adiponectina, la producción de NO y mejora la función endotelial58.
Se sabe que el ácido retinoico, derivado de la vitamina A, inhibe la expresión del gen de leptina en el tejido adiposo de ratones59. Sin embargo, en cultivos de sincitiotrofoblastos hay un efecto estimulador sobre la expresión y secreción de leptina60. En el tejido adiposo (blanco de su acción), el suministro de retinoides eleva la expresión de la proteína desacopladora de la mitocondria UCP-1, y esto se agrega a la reducción del exceso de tejido adiposo61.
CONCLUSIONES
Es probable que ciertas alteraciones durante el desarrollo fetal, algunas condiciones en el embarazo y las ECNT en el adulto, compartan mecanismos endógenos similares que pueden ser generados y/o regulados por factores externos.
Los procesos fisiopatológicos como EO, disfunción endotelial y mitocondrial y alteración en los niveles de adipoquinas, podrían asociarse con la programación fetal de ECNT. Es necesario establecer si las intervenciones como la complementación con micronutrientes y el EF, durante la gestación, pueden modular de forma benéfica una programación dañina para establecer estrategias en la prevención temprana de las ECNT.
AGRADECIMIENTOS
A COLCIENCIAS por el apoyo prestado a Robinson Ramírez-Vélez y a Isabella Echeverry, dentro del Programa de Doctorados Nacionales.
REFERENCIAS
1. Pinheiro AR, Salvucci ID, Aguila MB, Mandarim-de-Lacerda CA. Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin Sci. (Lond) 2008; 114: 381-92.
2. Kuh D, Ben-Shlomo Y. A life Course Approach to Chronic Disease Epidemiology. New York: Oxford University Press; 2004.
3. Zhang WZ, Venardos K, Chin-Dusting J, Kaye DM. Adverse effects of cigarette smoke on NO bioavailability: role of arginine metabolism and oxidative stress. Hypertension. 2006; 48: 278-85.
4. Nelson ME, Rejeski WJ, Blair SN, Duncan PW, Judge JO, King AC et al. Physical activity and public health in older adults: recommendation from the American College of Sports Medicine and the American Heart Association. Circulation. 2007; 28: 1094-105.
5. Koklu E, Akcakus M, Narin F, Saraymen R. The relationship between birth weight, oxidative stress and bone mineral status in newborn infants. J Paediatr Child Health. 2007; 43: 667-72.
6. Lowe DT. Nitric oxide disfunction in the pathophysiology of preeclampsia. Nitric oxide. Biol Chem. 2000; 4: 441-58.
7. Dörner G, Plagemann A, Neu A, Rosenbauer J. Gestational diabetes as possible risk factor for type I childhood-onset diabetes in the offspring. NeuroendocrinologyLett. 2000; 21: 355-9.
8. Barker DJ, Osmond C, Forsen TJ, Kajantie E, Eriksson JG. Maternal and social origins of hypertension. Hypertension. 2007; 50: 565-71.
9. Leon DA, Lithell H, Vagero D, McKeigue P, Koupilova I, Mohsen R et al. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15000 Swedish men and women born 191529. BMJ. 1998; 317: 2415.
10. Bertram CE, Hanson MA. Animal models and programming of the metabolic syndrome. Br Med Bull. 2001; 60: 103-21.
11. Luo ZC, Fraser WD, Julien P, Deal CL, Audibert F, Smith GN et al. Tracing the origins of «fetal origins» of adult diseases: Programming by oxidative stress? Med Hypotheses. 2006; 66: 38-44.
12. Gupta P, Narang M, Banerjee BD, Basu S. Oxidative stress in term small for gestational age neonates born to undernourished mothers: a case control study. BMC Pediatrics. 2004; 4: 14.
13. Longini M, Perrone S, Kenanidis A, Vezzosi P, Marzocchi B, Petraglia F et al. Isoprostanes in amniotic fluid: A predictive marker for fetal growth restriction in pregnancy. Free RadBiol Med. 2005; 38: 1537-41.
14. Franco Mdo C, Akamine EH, Di Marco GS, Casarini DE, Fortes ZB, Tostes RC et al. NADPH oxidase and enhanced superoxide generation in intrauterine undernourished rats: involvement of the renin-angiotensin system. Cardiovasc Res. 2003; 59: 767-75.
15. Lewis RM, Poore KR, Godfrey KM. The role of the placenta in the developmental origins of health and disease. Implications for practice. Rev Gynaecol Perinat Pract. 2006; 6: 70-9.
16. Ogita H, Liao JK. Endothelial function and oxidative stress. Endothelium. 2004; 11: 123-32.
17. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007; 115: 1285-95.
18. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004; 109: 27-32.
19. Savvidou MD, Hingorani AD, Tsikas D, Frölich JC, Vallance P, Nicolaides KH. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet. 2003; 361: 1511-17.
20. San Martin R, Sobrevia L. Gestational diabetes and the adenosine/L-arginine/nitric oxide (ALANO) pathway in human umbilical vein endothelium. Placenta. 2006; 27: 1-10.
21. Franco MCP, Arruda RMP, Dantas APV, Mitiko Kawamoto E, Fortes ZB, Scavone C et al. Intrauterine undernutrition: expression and activity of the endothelial nitric oxide synthase in male and female adult offspring. Cardiovasc Res. 2002; 56: 145-53.
22. Andersen MR, Walker LR, Stender S. Reduced endothelial nitric oxide synthase activity and concentration in fetal umbilical veins from maternal cigarette smokers. Am J Obstetr Gynecol. 2004; 191: 346-51.
23. Wang Y, Gu Y, Zhang Y, Lewis DF. Evidence of endothelial dysfunction in preeclampsia: Decreased endothelial nitric oxide synthase expression is associated with increased cell permeability in endothelial cells from preeclampsia. Am J Obstetr Gynecol. 2004; 190: 817-24.
24. Goodfellow J, Bellamy MF, Gorman ST, Brownlee M, Ramsey MW, Lewis MJ et al. Endothelial function is impaired in fit young adults of low birth weight. Cardiovasc Res. 1998; 40: 600-6.
25. López-Jaramillo P, Silva SY, Rodríguez-Salamanca N, Durán A, Mosquera W, Castillo V. Are nutrition-induced epigenetic changes the link between socioeconomic pathology and cardiovascular diseases? Am J Ther. 2008; 15: 362-72.
26. Sparre T, Reusens B, Cherif H, Larsen MR, Roepstorff P, Fey SJ et al. Intrauterine programming of fetal islet gene expression in rats-effects of maternal protein restriction during gestation revealed by proteome analysis. Diabetologia. 2003; 46: 1497-511.
27. Lee YY, Park KS, Pak YK, Lee HK. The role of mitochondrial DNA in the development of type 2 diabetes caused by fetal malnutrition. J NutrBiochem. 2005; 16: 195-204.
28. Hayakawa T, Noda M, Yasuda K, Yorifuji H, Taniguchi S, Miwa I et al. Ethidium bromide-induced inhibition of mitochondrial gene transcription suppresses glucose-stimulated insulin release in the mouse pancreatic beta-cell line betaHC9. J Biol Chem. 1998; 273: 20300-7.
29. Lee HK, Song JH, Shin CS, Park DJ, Park KS, Lee KU et al. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. 1998; 42: 161-7.
30. Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol. 2007; 83: 84-92.
31. Gualillo O, González-Juanatey JR, Lago F. The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. TCM. 2007; 17: 275-83.
32. Konukoglu D, Serin O, Turhan MS. Plasma leptin and its relationship with lipid peroxidation and nitric oxide in obese female patients with or without hypertension. Arch Med Res. 2006; 37: 602-6.
33. Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzmán M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001; 276: 25096-100.
34. Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. A. J Biol Chem. 2003; 278: 45021-6.
35. Dunger D, Ong K. Abundance of adiponectin in the newborn. Clin Endocrinol. 2004; 61: 416-7.
36. Taylor PD, Poston L. Developmental programming of obesity in mammals. Exp Physiol. 2007; 92: 287-98.
37. McMillen IC, Muhlhausler BS, Duffield JA, Yuen BSJ. Prenatal programming of postnatal obesity: fetal nutrition and the regulation of leptin synthesis and secretion before birth. Proc Nutr Soc. 2004; 63: 405-12.
38. J Ren. Leptin and hyperleptinemia - from friend to foe for cardiovascular function. J Endocrinol. 2004; 181: 1-10.
39. Wolfe LA, Weissgerber TL. Clinical physiology of exercise in pregnancy: a literature review. J Obstet Gynaecol Can. 2003; 25: 473-83.
40. Hegaard HK, Pedersen BK, Nielsen BB, Damm P. Leisure time physical activity during pregnancy and impact on gestational diabetes mellitus, pre-eclampsia, preterm delivery and birth weight: a review. Acta Obstet Gynecol Scand. 2007; 86: 1290-6.
41. Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007; 25: 2379-93.
42. Hood DA. Invited Rewiev: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001; 90: 1137-57.
43. Roberts JM, Lain KY. Recent insights into the pathogenesis of pre-eclampsia. Placenta. 2002; 23: 359-72.
44. Bonzini M, Coggon D, Palmer KT. Risk of prematurity, low birthweight and pre-eclampsia in relation to working hours and physical activities: a systematic review. Occup Environ Med. 2007; 64: 228-43.
45. Covas MI, R Elosua, M Fito, M Alcantara, L Coca, J Marrugat. Relationship between physical activity and oxidative stress biomarkers in women. Med Sci Sports Exerc. 2002; 34: 814-9.
46. You T, Nicklas BJ. Effects of exercise on adipokines and the metabolic syndrome. Curr Diab Rep. 2008; 8: 7-11.
47. Venu L, Harishankar N, Krishna TP, Raghunath M. Does maternal dietary mineral restriction per se predispose the offspring to insulin resistance? Eur J Endocrinol. 2004; 151: 287-94.
48. Venu L, Vijaya BN, Rajender RK, Raghunath M. Effect of maternal vitamin and mineral restrictions on the body fat content and adipocytokine levels of WNIN rat offspring. Nutr Metab. 2007; 4: 21-6.
49. DeVos L, Chanson A, Liu Z, Ciappio ED, Parnell LD, Mason JB et al. Associations between single nucleotide polymorphisms in folate uptake and metabolizing genes with blood folate, homocysteine, and DNA uracil concentrations. Am J Clin Nutr. 2008; 88: 1149-58.
50. Osrin D, Vaidya A, Shrestha Y, Baniya RB, Manandhar DS, Adhikari RK et al. Effects of antenatal multiple micronutrient supplementation on birthweight and gestational duration in Nepal: double-blind, randomised controlled trial. Lancet. 2005; 365: 955-62.
51. Ryan KA, Mitchell R, Shoemaker JD, Shay NF. Analysis of serum leptin levels in adult males during experimental zinc deficiency and repletion. FASEB J 1999; 13: 448.
52. Ott ES, Shay NF. Zinc deficiency reduces leptin gene expression and leptin secretion in rat adipocytes. Exp Biol Med. 2001; 226: 841-6.
53. Bagchi D, Vuchetich PJ, Bagchi M, Tran MX, Krohn RL, Ray SD et al. Protective effects of Zn salts on TPA-induced hepatic and brain lipid peroxidation, glutathione depletion, DNA damage and peritoneal macrophage activation in mice. Gen Pharmacol. 1998; 30: 43-50.
54. Venu L, Kishore YD, Raghunath M. Maternal and perinatal magnesium restriction predisposes the rat pups to insulin resistance and glucose intolerance. J Nutr. 2005; 135: 1353-8.
55. Sun X, Zemel MB. Dietary calcium regulates ROS production in aP2-agouti transgenic mice on high-fat/high-sucrose diets. Int J Obesity. (Lond) 2006; 30: 1-6.
56. Sun X, Zemel MB. Calcium and 1,25- dihydroxyvitamin D3 regulation of adipokine expression. Obesity. 2007; 15: 340-8.
57. Benavente CA, Jacobson EL. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Rad Biol Med. 2008; 44: 527-37.
58. Thoenes M, Oguchi A, Nagamia S, Vaccari CS, Hammoud R, Umpierrez G et al. The effects of extended-release niacin on carotid intimal media thickness, endothelial function and inflammatory markers in patients with the metabolic syndrome. Int J Clin Pract. 2007; 61: 1942-8.
59. Felipe F, Mercader J, Ribot J, Palou A, Bonet ML. Effects of retinoic acid administration and dietary vitamin A supplementation on leptin expression in mice: lack of correlation with changes of adipose tissue mass and food intake. Biochim Biophys Acta. 2005; 1740: 258-65.
60. Guibourdenche J, Tarrade A, Laurendeau I, Rochette-Egly C, Chambon P, Vidaud M et al. Retinoids stimulate leptin synthesis and secretion in human syncytiotrophoblast. J Clin Endocrinol Metab. 2000; 85: 2550-5.
61. Mercader J, Ribot J, Murano I, Felipe F, Cinti S, Bonet ML et al. Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology. 2006; 147: 5325-32.