Aplication of authenticity criteria in mitochondrial studies on archaic bone remains from a prehispanic Muisca population
Nancy Patricia Jara, MSc1, Mónica Díaz, MSc1, Victoria Villegas MSc2, Clara López de Mesa, Est1, Diana Torres, PhD3, Jaime Bernal, MD , PhD4, Alberto Gómez, PhD5, Ignacio Briceño, MD, PhD6
1. Professor researcher, Faculty of Medicine, Universidad de La Sabana, Chía, Colombia.
e-mail: nancy.jara@unisabana.edu.co monica.diaz1@unisabana.edu.co clara.lopez@unisabana.edu.co
2. Assistant Professor, Faculty of Natural Sciences and Mathematics, Universidad del Rosario, Bogotá, DC, Colombia.
e-mail: VictoriaVillegas@javeriana.edu.co
3. Associate Professor, Faculty of Medicine, Pontificia Universidad Javeriana, Bogotá, DC, Colombia.
e-mail: dmtorres@javeriana.edu.co
4. Director, Institute of Human Genetics, Pontificia Universidad Javeriana, Bogotá, DC, Colombia.
e-mail: jebernal@javeriana.edu.co
5. Professor, Faculty of Medicine, Pontificia Universidad Javeriana, Bogotá, DC, Colombia. e-mail: agomez@javeriana.edu.co
6. Professor researcher, Faculty of Medicine, Universidad de La Sabana, Chía, Colombia, and Associate Professor, Pontificia Universidad Javeriana, Bogotá, DC, Colombia. e-mail: ignaciobb@unisabana.edu.co
Received for publication September 15, 2009 Accepted for publication May 11, 2010
SUMMARY
Introduction: Ancient DNA (aDNA)studies can support hypotheses regarding ancient populations; molecular studies can analyze the local population’s genetic composition, minimizing biases introduced by later migrations, demographic expansions, mutations, and bottleneck effects. These analyses must be performed with special care because of the low DNA concentrations and contamination risk; therefore, it is necessary to establish protocols to guarantee the reproducibility and veracity of results.
Objective: The present study aims to establish a protocol to obtain ancient DNA from 16 pre-Columbian bone samples found in an excavation performed in the area «Candelaria La Nueva» in Bogota, Colombia, dated in the period «Muisca Tardio».
Methods: Four founder mitochondrial DNA Amerindian haplotypes were analyzed by high resolution restriction enzyme analyses, obtaining fragments between 121 and 186 base pairs (bp). Different analyses were performed following a strict control of authenticity criteria regarding laboratory conditions, including: positive and negative controls, reproducibility of results, and verification of particular characteristics present in ancient DNA.
Results: Results obtained from the bone samples showed the exclusive presence of haplogroup A in the population studied. This data support the statement of the archaeologists of a single biological population in space and time. The distribution of this haplogroup in a 100% frequency supports the hypothesis of Chibcha genetic affiliation.
Conclusion: The present study is a contribution to the study of genetic diversity in archaic American populations, allowing the integration of geographic and historic data with genetic characterization techniques associated with linguistic, ethnographic, and glottochronology patterns. Following the protocol proposed in the present study allows fulfilling authenticity criteria for ancient samples with the available techniques.
Keywords: Mitochondrial DNA; Archaic DNA; Muisca; Chibcha.
Aplicación de criterios de autenticidad en estudios mitocondriales en restos óseos de un grupo poblacional muisca prehispánico
RESUMEN
Introducción: Los estudios de ADN arcaico (aADN) pueden verificar hipótesis sobre las poblaciones del pasado, permiten analizar la composición genética, pudiéndose así soslayar sesgos introducidos por migraciones, expansiones demográficas, mutaciones y cuellos de botella acontecidos con posterioridad. Los métodos de análisis son objeto de cuidadoso procedimiento debido a la dificultad de recuperación y a la posibilidad de contaminación, por lo tanto es necesario establecer protocolos que garanticen la reproducibilidad y veracidad de los resultados.
Objetivo: El presente estudio tiene como fin establecer un protocolo para la obtención de ADN arcaico en 16 muestras óseas precolombinas encontradas en una excavación del sector de Candelaria La Nueva, Bogotá, Colombia, fechadas dentro del período Muisca Tardío.
Métodos: Se estudiaron 4 haplogrupos fundadores amerindios presentes en el ADN mitocondrial arcaico, mediante enzimas de restricción con base en estudios de alta resolución obteniendo fragmentos de tamaños entre 121 y 186 pares de bases (pb). Los distintos análisis se realizaron observando un estricto control de los criterios de autenticidad en relación con las condiciones de laboratorio, incluyendo el uso de controles y blancos, la reproducibilidad de resultados y la verificación de las características particulares que presenta el ADN arcaico.
Resultados: Los resultados obtenidos de las muestras óseas arcaicas establecieron la presencia única del haplogrupo «A» en la población estudiada. Estos datos apoyan la afirmación de los arqueólogos en cuanto a que se trata de una misma población biológica tanto espacial como temporalmente. La distribución de este haplogrupo en una frecuencia del 100% soporta la filiación genética con los grupos chibcha.
Conclusión: Este trabajo es una contribución al estudio de la diversidad genética en las poblaciones americanas antiguas, permitiendo abordar la integración de datos geográficos e históricos con técnicas de caracterización genética asociados con patrones lingüísticos, etnogeográficos y glotocronológicos.
Palabras clave: ADN mitocondrial; ADN arcaico; Muisca; Chibcha.
Current population studies seek to infer genomic evolutionary processes, possibly introducing bias due to migration, population expansion, bottlenecks, and mutations producing allele frequency changes in different human populations. The analysis of archaic DNA (aDNA), for the DNA recovered from biological samples that have not been specifically preserved for further analysis, allows going back in the history of populations, establishing the genetic variability and verifying hypotheses from other disciplines. Thus, aDNA analysis has permitted studying the origins of different populations without the risks of the projections elaborated from contemporary populations1.
When conducting ancient DNA studies, it is important to know the postmortem biological degradation process to understand the characteristics of the DNA studied and the degree of conservation of samples to be studied. Broadly speaking, after death there is no supply of oxygen to tissues, causing a failure in cellular metabolism and repair mechanisms. Therefore, the biological molecules begin to be degraded by endonucleases from cell membranes. After this process, soft tissue decomposition is followed by rapid growth of intestinal bacteria; subsequently, involving the action of soil microorganisms, fungi, plants, insects, and scavengers. Likewise, the postmortem degradation rate depends on environmental factors like temperature, pH, oxygen supply, and the presence of water. In advanced decomposition stages, the soft tissues disappear almost completely, while the appendages of vertebrates do not lose their organic components. Similarly, hard tissues like bones and teeth are preserved even under ordinary conditions, due to the low amount of water and low enzyme content and to the presence of hydroxyapatite mesh that holds and protects the DNA2.
DNA is considered a relatively unstable molecule that can be degraded rapidly. However, there are theoretical and empirical grounds to suggest that the maximum survival of ancient DNA is 50.000 to 1.000.000 years. It has been determined that the most common type of damage in ancient DNA is degradation into small fragments, usually between 100-500 bp; these conditions result from enzymatic processes that occur postmortem, such as microorganism action, non-enzymatic hydrolytic cleavage of phosphodiester, and glycosidic bonds and the action of free radicals3. But, the aDNA degradation percentage depends on its preservation conditions and can vary even among museum specimens of the same age4.
On the other hand, it is important to consider the likelihood of greater preservation of mtDNA and its number of copies (100 to 1000 copies) per cell in contrast to the single copy of nuclear DNA, so it is better to use this genome in aDNA studies. The mtDNA is composed of two basic regions: the hypervariable region also known as D-loop or control region, which corresponds to the non-coding segment of mtDNA, and the coding region, which corresponds to 94% of the genome, located almost entirely in the H chain, with 28 genes in a very compact structure5.
Currently, there are two main molecular methods used to optimize the study of mitochondrial genome and human variability: analysis of restriction enzyme polymorphisms (RFLPs) and sequencing of the hypervariable region of the control region. These two methods allow the study of haplogroups present in a population, as well as haplotypes associated with specific polymorphisms based on phylogenetic analysis. The data obtained from these tests has been used as evidence to support hypotheses on timing, origin, size, and number of migrations from Asia to America, and have allowed studying the demographic history of these regions6.
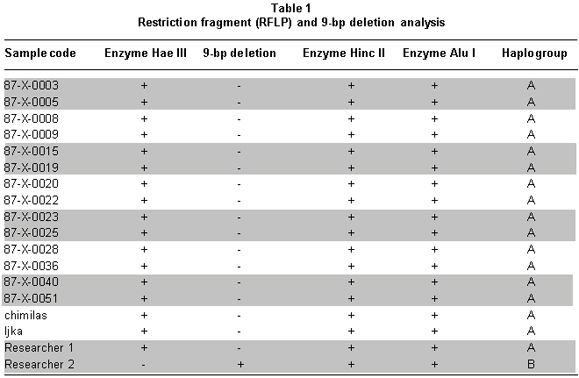
Through the study of RFLPs, we can analyze combinations of the presence or absence of genetic markers in the different restriction sites and a nine base pair (bp) deletion that defines founder Native American Haplogroups A, B, C, D, and X, also found in Asia and Siberia, establishing phylogenetic relationships among them7. Restriction site gain with the HaeIII enzyme at position 663 corresponds to haplogroup A, the loss of a restriction site with the HincII enzyme at position 13259 corresponds to haplogroup C, the loss of a restriction site with the AluI enzyme at nucleotide 5176 corresponds to haplogroup D, and the deletion of nine base pairs in the COII/tARNlys region corresponds to haplogroup B8.
Another method to assess mtDNA variation includes direct sequencing of hypervariable segments of the control region, located between transfer RNA genes for proline (tARNpro) and transfer RNA to the amino acid phenylalanine (tARNphe), which accumulates mutations faster than elsewhere in the mtDNA, and determines a mitochondrial haplogroup9 for each different sequence.
Considering the difficulties in analyzing ancient DNA and the high probability of contamination in the research made in this field, some aspects in handling material containing ancient DNA should be taken into account to establish consensus for essential requirements in the validation of results; these have been called «criteria of authenticity» for ancient DNA10-14.
These criteria include aspects such as the preservation of samples obtained by maintaining a chain of custody and handling always in optimal sterile conditions, from the collection site to its final destination in the laboratory. Also, the individuals who handle the samples should have personal protection to avoid contamination with foreign contemporary DNA, and researchers should be typed to exclude the possibility of their DNA being amplified. In addition, the laboratory should be disinfected and cleaned each time it is used for the analysis of ancient DNA samples, and strict precautions must be taken in the preparation and management of all solutions to avoid contamination with aerosols produced by previous DNA amplification in other samples. Sterile cubicles and PCR areas for aDNA should be areas isolated from modern DNA work.
Additionally, in processing fragments of ancient DNA, blanks need to be made, positive and negative controls to assure sample quality. It is important to repeat the extraction and amplification from the same or different extracts from the same sample, at least in two different laboratories, performed by different personnel to detect the presence of foreign DNA. Amplification of short fragments of ancient DNA (no more than 500 pb) is the rule.
With these types of protocols, phylogenetic studies are possible, especially, in extinct species, although these criteria are not necessariy easy to apply in human populations, polymorphisms known and defined as native American can be analyzed. However, it is important to have a preliminary database on the polymorphisms under study, which includes the frequency and geographical distribution of each polymorphism reported to have a reference DNA sequence. Finally, biochemical testing, quantification, sequencing, and cloning of the amplification products and the genetic determination of gender are considered additional criteria of authenticity, and these complementary protocols are only performed when the amount of aDNA is sufficient.
MATERIALS AND METHODS
A retrospective analytical study of archaic bones was performed on 24 individuals belonging to a reference collection of skeletal material from the Instituto Colombiano de Antropología e Historia de Colombia (ICANH) kept in the Genetics Institute laboratory at Pontificia Universidad Javeriana in Bogotá. These archeological samples met the standards established by the inclusion criteria. The rescue was carried out between August 23 and September 22, 1987. The cultural origin of this finding was established based on ceramics, the morphology of the tombs, and dwellings by archaeologists from the ICANH and further tests on isotopes d13C, d15C, and d15N were made at Geochron Laboratories in Cambridge, Massachusetts. Based on these criteria, it appeared that the bone material recovered belongs to the late Muisca period, between 1175 and 1250 AD15.
The criteria for this study included the certification of origin by the ICANH, the month and year of the bone sample ingress to ICANH; the number of the skeleton and the physical conditions of each bone were certified by an anthropologist. Subsequently, 16 samples were selected as meeting the established selection criteria15.
Cleaning, soaking, deproteinization, extraction, and re-extraction of DNA from bone. Fragments of archaic and modern bones (positive control current mitochondrial DNA) were polished with sterile sandpaper (number 300) to eliminate contamination from foreign DNA, according to the standard protocol by Kalmar et al.16 and cleaned with 5.2% sodium hypochlorite (NaOCl2) to remove contaminant DNA belonging to individuals who handled the samples before extraction procedures, as suggested by Kemp et al.17
After this, we macerated dry bone fragments (approximately 6 cm long), which were wrapped in sterile aluminum foil. For this procedure, we used a hammer (hammer weight 250 g) previously disinfected with sodium 5.2% hypochloride and 70% ethanol to avoid contamination between each sample. The powder obtained with an average weight of 20 mg was transferred to a sterile 15-ml tube (Falcon®), and then 4.7 ml of cell lysis solution (0.5M EDTA, 10% SDS, pH 8.0) was added and briskly stirred. Then we added 400 ml of Proteinase K (1 mg/ml) per sample and mixed by inversion. Samples were stirred on a rotating platform for 24 h at 37°C and then on a serological bath at 55°C for 24 hours. After 48 h of agitation, the samples were centrifuged at 3000 rpm for 5 minutes and the supernatant (4 ml approximately) was transferred to a new sterile 15 ml Falcon® tube.
Protein precipitating solution (900 ml) was added to each sample, (10 M ammonium acetate) and mixed by inversion for 5 minutes and the samples were taken to the freezer (-20oC) for 5 minutes. New centrifugation was performed at 4000 rpm for 20 minutes. The supernatant was extracted and the previous step was repeated to avoid possible artifacts that affect the quality of extracted DNA. Each sample was supplemented with 5 ml of 90% isopropanol at -20°C, gently shaken by inversion for 5 minutes, and then the samples were brought to -20°C for 15 minutes, centrifuged at 5000 rpm for 30 minutes, and the supernatant was discarded and each tube was incubated, inverted on absorbent paper at 37°C for 1 h. The sediment was resuspended rehydrated with 200 ìl of water and was left stirring overnight at 37°C to preserve the good quality of DNA16 obtained. Subsequently, DNA was electrophoresed on 1.0% agarose at 150 V for 45 minutes. To visualize the DNA on each extracted sample, the gel was colored with ethidium bromide and visualized on a UV transilluminator at 312 nm. Images were obtained with a Polaroid camera (Fotodyne 5®).
To assure the removal of most PCR inhibitors, a new extraction was conducted on the DNA of extracted samples (re-extraction) following the protocol of a commercial saline extraction kit (Corpogen DNA 2000® Reference BM-00).
To remove most PCR inhibitors, we conducted a new extraction on the DNA of extracted samples (re-extraction) following the protocol of a commercial saline extraction kit (Corpogen DNA 2000® Reference BM-00).
Afterwards, a volume of 400 ml of deproteinization solution was added, and the tubes were frozen at a temperature of -20°C for 15 minutes, and then centrifuged at 13000 rpm for 5 minutes. A total of 90% isopropanol was added at 4°C to each tube in a proportion corresponding to 1/6 of the total volume, and the tubes were centrifuged at 13000 rpm for 10 minutes. The supernatant was discarded and tubes were left to dry inverted on absorbent paper at 37°C for 1 h. Then, the resuspended sediment from each sample on 100 ml of deionized water was taken to a shaking platform overnight at 37°C. Finally, the re-extracted samples were electrophoresed on 1% agarose at 150 V for 45 minutes to visualize the corresponding DNA. Images were obtained with a Polaroid camera (Fotodyne 5®).
DNA amplification. Amplification conditions were conducted according to the protocol suggested by Kalmar et al.16 The reactions were carried out in the thermocycler I Cycler® (BioRad). The program for amplification was: initial denaturation: 97°C for 5 minutes, and repeated 35 cycles: denaturation at 95°C for 1 minute, annealing as the primer used (for HaeIII at 60°C, for HincII at 53°C, at 56°C for AluI and 9bp deletion at 52°C for 1 minute, extension at 72°C for 1.5 minutes, continuing with final extension at 72°C for 5 minutes. We used Taq DNA Polymerase Recombinant from Invitrogen®. The amplifications were carried to a final volume of 25 ml; each sample required a specific volume of DNA, 2, 2.5 or 3 ml. The primers used to determine each Haplogroup were:
.Haplogroup A: HaeIII A663G L635: tgaaaat gtttagacggcctcacatc, H708: tagagggtgaactcactggaac, with a fragment length of 121 bp;
.Haplogroup B: deletion of 9 bp acagtttcatgcccatcgtc L8215, H8297: ctgtaaagctaacttagcat. With a 112-bp fragment length
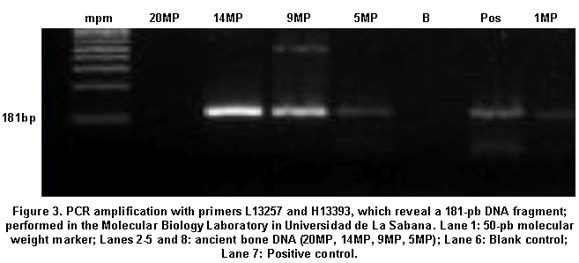
.Haplogroup C: A13263G L132057: aatcgtagcctt ctccacttca, H13393: gaacaagatattcgaaaaatagga, a 181-bp fragment length;
.Haplogroup D: L5054 C5178A: taggatgaataata gcagctctaccg, H5189: acacccttaattccatccaccc with a fragment length of 183 pb18.
Analysis with restriction enzymes. With the product of the amplification and/or re-amplification of the samples, we carried out a DNA digestion with restriction enzymes AluIII, HincII and HaeIII. In each case, 12 ml of each PCR product was taken and 2 ml of restriction enzyme were added with 2 ml of 10x buffer, completed with deionized water to a final volume of 20 ml. This mixture was left overnight at 37°C. Additionally, only in the case of the HincII enzyme, we added 0.5 ml of bovine serum albumin (BSA). In order to visualize the fragments generated by the restriction enzymes, we used a 2% agarose gel and 12% poliacrilamide gel electrophoresis (PAGE), which had a better resolution especially for the samples with small amounts of DNA. We performed a 30-minute preliminary electrophoresis at 100 volts. Thereafter, the samples were placed on the gel and run at 100 volts for 40 minutes, and then at 150 volts for 120 minutes. Then we proceeded with an ethidium bromide staining and photographed the stained gel with a Polaroid camera (Fotodyne 5®).
Detection of the 9-bp deletion. In order to detect the 9bp deletion, the amplification product of each sample was run on a 2.5% agarose gel and then on a 12% polyacrylamide gel as described.
This research followed the protocols from the Laboratory of the Genetics Institute in Pontificia Universidad Javeriana (Bogota) and the Molecular Biology laboratory in Universidad de La Sabana (Chia).
RESULTS
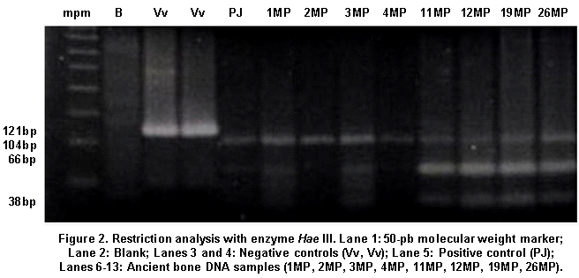
A total of 16 aDNA samples were analyzed. In all, 13 different individuals were studied and three of them were studied in duplicate for two purposes: comparison of the duplicated results to determine the quality of the procedure and identification of possible contamination with exogenous DNA, based on the fulfillment of the criteria of authenticity. Amerindian Haplogroup A was identified in all bone samples by means of RFLPs. Control samples of contemporary Indians also belonged to haplogroup A. Control samples of the researchers who came into direct contact with the samples belonged, respectively, to haplogroup A (investigator 1) and to haplogroup B (investigator 2) (Figures 1, 2, and 3). Also, 100% of the modern subjects’ DNA (Chibcha) corresponds to haplogroup A.
Additionally, investigator 2 in the same laboratory of the Genetics Institute in Pontificia Universidad Javeriana took a group of samples and performed various protocols used in this study, obtaining the same results (Table 1). Finally, we took some of the archaic and modern samples and processed them at the Molecular Biology Laboratory in Universidad de La Sabana, where standardized protocols were applied (Figure 3). The results were identical to those achieved by the two researchers in the laboratory of the Genetics Institute in Pontificia Universidad Javeriana, indicating the reproducibility by different researchers in different laboratories.
DISCUSSION
In this study, we performed a detailed analysis of the main aspects that should be taken into account in handling human remains subsequently undergoing archaic mtDNA analysis, assessing the implementation of the criteria of authenticity for ancient DNA analysis in a pre-Colombian Chibcha population, from an indigenous burial ground located southwest of Bogota, at the site of «Candelaria La Nueva».
This tool is presented as a new possibility of human mtDNA analysis, because it allows identifying the pedigree of the individuals from the archaeological site in this pre-Hispanic community with a high level of reliability to create a standard to compare with similar studies of the pre-Hispanic period.
The data obtained from the analysis of samples made via ancient mtDNA polymorphisms in a subpopulation in the sector of «Candelaria La Nueva», Bogotá, Colombia, showed the mitochondrial haplogroup ‘A’, which is usually dominant on Chibcha groups, as reported by Melton et al.6 on Amerindians of Central and South America, and Guardado-Estrada et al.20 in a Mexican population. This supports the archaeological findings previously obtained by linguists and anthropologists who classified this population as belonging to the Chibcha-speaking group15. It is important to note that bones coming from this archaeological finding were in good condition, facilitating their study.
The study is based on the criteria of authenticity for ancient DNA analysis to detect foreign DNA contaminants, avoid false positives, and give veracity to the results obtained. One of the most common problems in such studies is the modern DNA contamination due to manipulation by the group of archaeologists. For this reason, stringent steps for cleaning the bone areas that could have been in contact with contaminating DNA were taken. During the excavation at the site «Candelaria La Nueva», anthropologists implemented the necessary bio-security measures. This material was inventoried, classified, and stored properly until the completion of the analysis of ancient DNA15,16.
Furthermore, the researchers were typed for mtDNA haplogroups, which allowed them to control and specify eventual contamination on the manipulated samples. Whenever the results matched bone samples, archaic and researchers, these same samples were analyzed by other researchers and other laboratories to confirm the results previously obtained. Similarly, to avoid contamination of foreign DNA in the laboratory, it was necessary to establish suitable conditions for disinfection and sterilization, and the floors and walls were cleaned at the beginning and end of work with 5% NaOCl2; work surfaces and equipment were also cleaned with 5% NaOCl2 and subsequently exposed to ultraviolet radiation10-14. It was also important to avoid sources of contamination of other samples used for DNA studies in the same laboratory; therefore, the areas of pre-and post-PCR were separated from each other, as well as from areas of modern DNA analysis. Also, preparation of the solutions required for the different protocols were made in an isolated space for ancient DNA processing and every precaution for their preparation was taken10-14.
For the genetic study of skeletal remains, each bone sample was split into four fragments worked in different laboratories to test the reproducibility of results11-14. We also implemented the use of deionized water as a blank control for every amplification and a master mix without DNA for re-amplification. To assure the obtention of expected results. Therefore, a low percentage of false positives were found during the initial standardization of the protocols, and contamination sources were detected and eliminated in subsequent procedures, as the blank control values were always negative. No contamination was observed in either of the two laboratories13. On the other hand, both modern DNA samples belonging to researches, and blood samples from indigenous populations in Colombia, which had already been typed were used as control. This allowed us to establish the differences among DNA from contemporary indigenous individuals and ancient Amerindian DNA.
In this study, we obtained fragments of 121, 183, and 187 bp, allowing the confirmation of the presence of archaic DNA in skeletal remains10-14. The fragments of 181 pb and 183 pb had to be amplified and re-amplified more times than those of 121 pb, which establishes an inverse relationship between size and efficiency of the PCR process, as fragments of 121 pb were more easily identified10-14.
Also, the results were consistent with those expected from the geographical distribution of the Amerindian groups studied, compared with the data provided in the banks of founding Amerindian haplogroups, where the frequency and geographical distribution of mtDNA polymorphisms are well established9.
Additionally, other authenticity criteria could not be considered in the present study because the amount of aDNA was not enough. All the protocols set out in this research were performed at the Molecular Biology Laboratory in Universidad de La Sabana, obtaining the same results as those obtained in the laboratory of the Genetics Institute in Pontificia Universidad Javeriana, implying that the results were reproducible.
This work established guidelines that serve as reference for further studies based on a similar methodology. Determination of dominant mitochondrial haplogroups in native Colombia establishes the relationships of ancient and modern populations with other studies and a clear impact on association studies of genetic markers.
ACKNOWLEDGEMENTS
We would like to recognize the strong commitment of Universidad de la Sabana and Pontificia Universidad Javeriana, and also the contributions of our colleagues in both institutions. In particular, we would like to acknowledge the logistics support from Dr. Fernando Lizcano, Director of the Molecular Biology Laboratory in Universidad de La Sabana. Finally, we would like to recognize the help of Dr. Enrique Bautista who donated the ancient bone samples to this investigation.
Conflict of interest. None of the authors has conflicts of interest related to this study.
REFERENCES
1. Kolman CJ, Tuross N. Ancient DNA analysis of human populations. Am J Phys Anthropol 2000; 11: 5-23.
2. Gilbert T, Willersslev E, Hansen A, Barnes I, Rudbec L, Lynnerup N, Cooper A. Distribution patterns of postmortem damage in human mitochondrial DNA. Am J Hum Genet. 2003; 72: 32-47.
3. Görerstromöm A, Collins M, Angerbjörn A, Liden, K. Bone preservation and DNA amplification. Archaeometry. 2002; 44: 395-404.
4. Moraga, M. Asprillaga E, Santero C, Standen V, Carvallo P, Rothhammer F. Análisis de ADN mitocondrial en momias del norte de Chile avala hipótesis de origen amazónico de poblaciones andinas. Rev Chil Hist Nat. 2001; 74: 1-12.
5. Brandon M, Lott M, Nguyen K, Spolim S, Navathe S, Baldi P. MITOMAP: a human mitochondrial genome database-2004 update. Nucl Acids Res. 2005; 3: 611-3.
6. Melton PE, Briceño I, Gómez A, Devor EJ, Bernal JE, Crawford MH. Biological relationship between Central and South American Chibchan speaking populations: evidence from DNA. Am J Phys Anthropol. 2007; 133: 753-70.
7. Schurr G. T. The peopling of the new world: perspectives from molecular anthropology. Ann Rev Anthropol . 2004; 33: 551- 83.
8. Ingman M, Gyllensten E. Analysis of the complete human mtDNA genome: methodology and inferences for human evolution. Am Gen Assoc. 2001; 92: 454-61.
9. Bandelt, HJ, Herrnstadt C, Yao, YG, Kong Q, Kivisild T, Rengo C et al. Identification of native American founder mtDNAs through the analysis of complete mtDNA sequences: some caveats. Ann Hum Gent. 2003; 67: 512-24.
10. Noonan JP, Coop G, Kudaravalli S, Smithe D, Krause J, Alessi J et al. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006; 314: 1113-8.
11. Kolman CJ, Tuross N. Ancient DNA analysis of human populations. Am J Phys Anthropol. 2000; 11: 5-23.
12. Pääbo S, Poinar H, Serre D, Jaenicke- Depres V, Hebler J, Kuch M et al. Genetic analysis from ancient DNA. Ann Rev Genet. 2004; 38: 645-79.
13. Yang D, Watt K. Contamination controls when preparing archaeological remains for ancient DNA analysis. J Archaeol Sci. 2004; 32: 331-36.
14. Adachi N, Umetsu K, Wataru T, Sakaue K. Phylogenetic analysis of the human ancient mitochondrial DNA. J Archaeol Sci 2004; 31: 1339-48.
15. Cifuentes, A, Moreno L. Proyecto de rescate arqueológico de la Avenida Villavicencio (Barrio Candelaria la Nueva). Bogotá, DC: Instituto Colombiano de Antropología; 1987.
16. Kalmár T, Bachrati CZ, Marcsik A, Raskó. A simple and efficient method for PCR amplifiable DNA extraction from ancient bones. Nucl Acid Res. 2000; 28: e67.
17. Kemp BM, Smith DG. Use of bleach to eliminate contaminating DNA from the surface of bones and teeth. Forensic Sci Int. 2005; 10: 53-61.
18. Wang S, Lewis C, Jacobsson M, Ramachandran S, Ray Nicole, Bedoya G et al. Genetic varation and population structure in native Americans. Plos Genet. 2007; 3: 2049-67.
19. Mesa N, Mondragón M, Soto I, Parra M, Duque C, Ortiz- Barrientos D et al. Autosomal, mtDNA, and Y-chromosome diversity in Amerinds: pre- and post-Columbian patterns of gene flow in South America. Am J Hum Gen. 2000; 67: 1277-86.
20. Guardado-Estrada M, Juárez-Torres E, Medina-Martínez I, Wegier A, Macías A, Gómez G et al. A great diversity of Amerindian mitochondrial DNA ancestry is present in the Mexican mestizo population. J Hum Genet. 2009; 54: 695-705.