18p-syndrome: Presentation of two cases with alobar holoprosencenphaly
Harry Pachajoa, MD, PhD (A)1, Wilmar Saldarriaga, MD, MSc2, Carolina Isaza, MD, MSc3
1. PhD student in Biomedical Sciences, Assistant Professor, School of Basic Medical Sciences, Universidad del Valle. Professor, Faculty of Health Sciences, Universidad Icesi, Cali, Colombia. e-mail: harrympl@yahoo.com
2. Assisting Professor, School of Basic Medical Sciences, School of Medicine, Universidad del Valle, Cali, Colombia.
e-mail: wsaldarriaga0608@yahoo.com
3. Full Professor, School of Basic Medical Sciences, Vice-rector of Research, Universidad del Valle, Cali, Colombia.
e-mail: carolinaisa@cable.net.co
Received for publication May 26, 2009 Accepted for publication August 18, 2009
SUMMARY
Introduction: The syndrome by deletion of the short arm of chromosome 18 is an infrequent syndrome, and its phenotypical variability makes it difficult to recognize. Its most frequently observed clinical characteristics include mental retardation, growth retardation, craniofacial malformations, including long ears, microcephaly and short neck; other less frequent associated malformations include holoprosencephaly.
Case report: We present two patients with deletion of the short arm of chromosome 18, one presented a de novo mutation and the other was produced by a balanced translocation 6p/18p of maternal origin. Both patients presented alobar holoprosencephaly and cebocephaly, low-frequency clinical characteristics in this syndrome.
Discussion: alobar holoprosencephaly is a malformation appearing in 10% of patients with deletion of the short arm of chromosome 18; we review the probable physiopathology of holoprosencephaly in this syndrome.
Keywords: Syndrome by deletion of short arm of chromosome 18; (18p-) syndrome; Alobar holoprosencephaly.
Síndrome 18p-: Presentación de dos casos con holoprosencefalia alobar
RESUMEN
Introducción: El síndrome por deleción del brazo corto del cromosoma 18, es un síndrome poco frecuente y su variabilidad fenotípica lo hace difícil de reconocer. Las características clínicas observadas con más frecuencia incluyen retardo mental y de crecimiento, malformaciones craneofaciales que incluyen orejas largas, microcefalia y cuello corto; otras malformaciones asociadas menos frecuentes incluyen la holoprosencefalia.
Reporte de casos: Se presentan dos pacientes con deleción del brazo corto del cromosoma 18, uno presentado de novo y otro producido por translocación balanceada 6p/18p de origen materno. Ambos pacientes presentaron holoprosencefalia alobar y cebocefalia, características clínicas de baja frecuencia en este síndrome.
Discusión: La holoprosencefalia alobar es una malformación que se presenta en 10% de los pacientes con deleción del brazo corto del cromosoma 18; se revisa la probable fisiopatología de la holoprosencefalia en este síndrome.
Palabras clave: Síndrome por deleción del brazo corto del cromosoma 18; Síndrome (18p-); Holoprosencefalia alobar.
The syndrome by deletion of the short arm of chromosome 18 (18p-) was first described by Grouchy et al. in 1963; it is a syndrome with an estimated prevalence of 1 in every 50,000 live births and its phenotypic variability makes it difficult to recognize; however, the most frequently observed clinical characteristics include mental retardation, growth retardation, low height, pectus excavatum, craniofacial malformations including long ears, ptosis, microcephaly, and short neck1. There are case reports of holoprosencephaly diagnosed prenatally2.
Herein, we furnish a report of two patients with deletion of the short arm of chromosome 18 (OMIM 146390) with alobar holoprosencephaly, given that this association is infrequent and there are not many reports of such in literature.
Case report 1
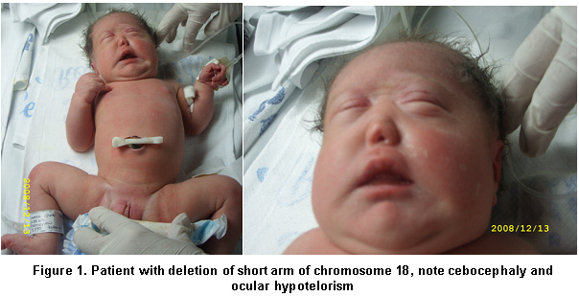
We present a female newborn, offspring of a 41-year-old mother and product of a third pregnancy. Prenatal ultrasound during the first, second, and third trimesters revealed ventriculomegaly and alobar holoprosencephaly. The birth took place at Hospital Universitario del Valle in the city of Cali, with an Apgar score of 8 and 9 on the first and fifth minute; placenta, umbilical cord and blood vessels without alteration, Ballard GA of 35 weeks, weight: 2535 g (p=3-25), height: 46 cm (p=10), head circumference: 32 cm (p=5), distance between inner edges: 16 mm (p<3), distance between outer edges: 60 mm (p=3-25). The physical exam found rounded face, sinophris, ocular hypotelorism, cebocephaly, single nostril, and micrognathia (Figure 1).
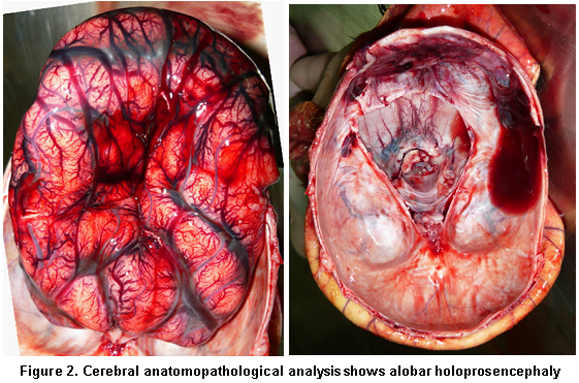
Paraclinical findings. Simple cerebral scan revealed a single ventricle, fused thalamus; findings compatible with alobar holoprosencephaly, echocardiogram: normal and renal ultrasound: normal. Cytogenetic findings (g-banding karyotype, 750-band resolution): 46, XX, of (18)(p11.2). The patient died one day after birth, with the anatopathological exam revealing the findings reported by the ultrasound and scan (Figure 2).
Case report 2
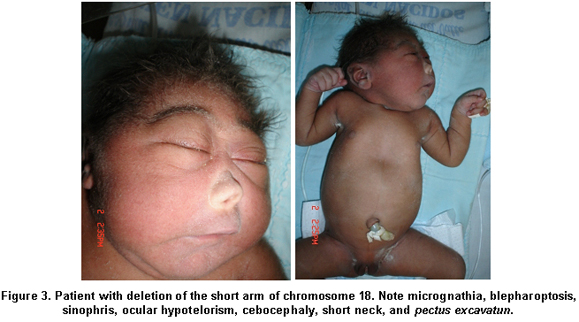
We present a female newborn, offspring of a primigest 17-year-old mother. Prenatal ultrasound was performed during the first, second, and third trimesters; the first two revealed no alterations and the third showed evidence of asymmetry of the ventricular system. The birth took place at Hospital Universitario del Valle in the city of Cali, with an Apgar score of 8 and 9 on the first and fifth minute; placenta, umbilical cord and blood vessels without alteration, Ballard GA of 38 weeks, weight: 2200 g (p=5), height: 47 cm (p=10), head circumference: 28 cm (p=5), distance between inner edges: 15 mm (p<3), distance between outer edges: 58 mm (p=3-25). The physical exam found fused sutures, posterior and anterior fontanelle <1 cm, rounded face, sinophris, ocular hypotelorism, cebocephaly, single nostril, micrognathia, flat osseous palate, and pectus excavatum (Figure 3).
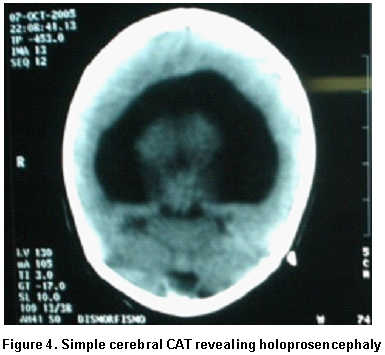
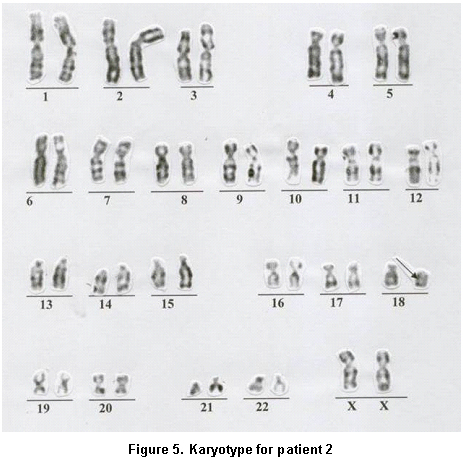
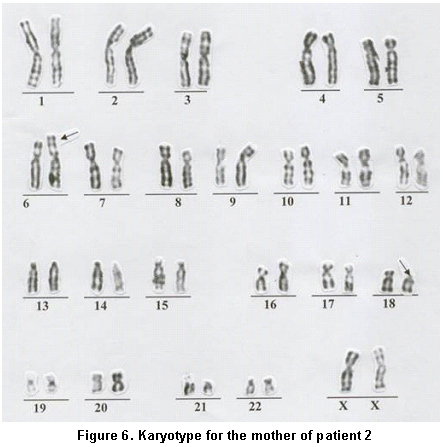
Paraclinical findings. Simple cerebral scan revealed a sole ventricle, fused thalamus, without evidence of structures in the medial line; alobar holoprosencephaly is concluded (Figure 4), echocardiogram: normal, renal ultrasound: normal. Cytogenetic findings (g-banding karyotype, 750-band resolution): 46, XX, of (18)(p11.2) see Figure 5, in the Karyotype the mother was found 46, XX, t (6p, 18p) (18pter�™18p11.2: 6p24�™6qter), der. 18 (Figure 6).
The mother is evaluated without finding visible malformations. The patient died 11 days after birth.
Ethics. In both cases, the parents signed an informed consent allowing photographs to be taken and retrieval of information from the clinical charts.
DISCUSSION
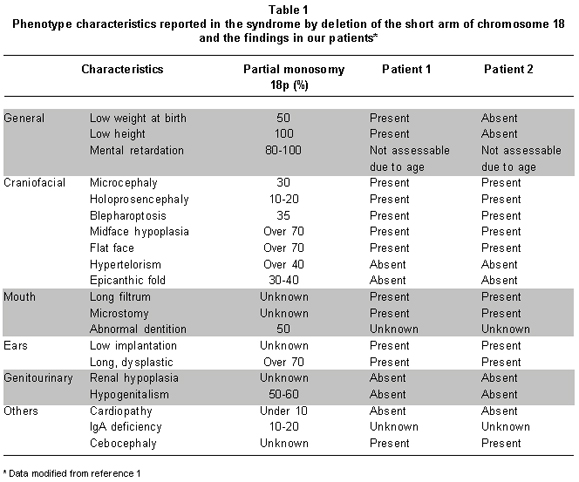
Nearly 150 cases have been reported with the syndrome by deletion of the short arm of chromosome 18, and 85% of the cases have originated in a de novo deletion and less frequently in familial origin1. In case 2, the newborn presents an 18p11.2 deletion, considered of maternal origin, because of a finding in the mother of a balanced translocation 6p/18p, a finding infrequently reported1. The clinical characteristics found in our cases are similar to those described by other authors (Table 1), although less frequent findings were found like alobar holoprosencephaly and the presence of a single nostril3.
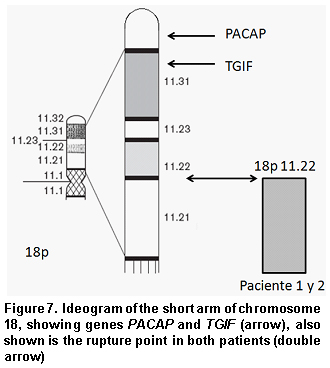
Approximately 50% of the cases with holoprosencephaly are associated to chromosome alterations or to a monogenic syndrome. This is the most severe malformation in patients with 18p- syndrome, which is present in close to 10% of the cases. The severity of the holoprosencephaly varies from cyclopy to facial malformations with a central incisor. Four genes for holoprosencephaly are known: SHH (7q36), ZIC2 (13q32), SIX3 (2p21), and TGIF mapped in the distal most part of the short arm of chromosome 18 (18p11.3)4. Such may be the one affected in our patients with a consequent abnormal cerebral formation and production of alobar holoprosencephaly (Figure 7).
The clinical characteristics or the gravity of the clinical condition are not related to the extension of the deletion of the short arm of chromosome 18; as can be noted by the description of the two cases reported by Hadedank5, the ratio between women and men is 3 to 2, and gonodal dysgenesis has been reported along with menstrual and fertility disorders1.
Among the clinical characteristics reported, there is also deficiency in the growth hormone, which may be regulated by a critical region located in the 18p distal region between 18p11.23 and 18pter. Analysis of this region has revealed it contains a gene that codifies for the pituitary adenylate cyclase-activating polypeptide (PACAP), mapped in the 18p11.32 region. PACAP and the growth release hormone (GRH) are part of the same super-family of regulatory neuropeptides and both have hypophysiotropic activity; low height is common in patients with Monosomy 18p and it may be a result of deficiency of the growth hormone secondary to an alteration in a path involving the genes mentioned, where PACAP would be absent because of the deletion4.
The prenatal diagnosis of this syndrome is possible through amniocentesis; in addition, an increase of nuchal translucency has been reported in this syndrome1.
Genetic advice for the parents should be done by a specialized team, including a geneticist physician, to estimate the risk of recurrence according to each case and especially in cases presented through translocations. Care of these patients should be carried out by an interdisciplinary group according to age and findings in each patient.
Conflict of interest. None of the authors has conflicts of interest related to this study.
REFERENCIAS
1. Turleau C. Monosomy 18p. Orphanet J Rare Dis. 2008; 19: 4.
2 . Wang J-CC, Nemana L, Kou SY, Habibian R, Hajianpour MJ. Molecular cytogenetic characterization of 18; 21 whole arm translocation associated with monosomy 18p. Am J Med Genet. 1997; 71: 463-6.
3. Zumel RM, Dranaude MT, Delicado A, Díaz de Bustamante A, de Torres ML, Lopez-Pajares I. The 18p- syndrome. Ann Genet. 1989; 32: 160-3.
4. Portnoi MF, Gruchy N, Marlin S, Finkel L, Denoyelle F, Dubourg C et al. Midline defects in deletion 18p syndrome: clinical and molecular characterization of three patients. Clin Dysmorphol. 2007; 16: 247-52.
5. Hadedank M, Trost-Brinkhues G. Monosomy 18p- and pure 18p- in a family with translocation (7, 18). J Med Genet. 1983; 20: 377-9.