HPV infection and carcinogenesis in the upper aero-digestive tract
Andrés Castillo, BSc, PhD*
* Professor, Department of Physiological Sciences, School of Health, Universidad del Valle, Cali, Colombia. e-mail: andrescg76@hotmail.com
Received for publication July 13, 2010 Accepted for publication November 17, 2010
SUMMARY
Recently, molecular and epidemiological studies have shown an etiologic role for human papillomavirus (HPV) in a subset of upper aero-digestive tract (UADT) cancers in women and men. Oral HPV infections acquired through oral sex seems to be the main risk factor for HPV-associated oral cancers. The high-risk type HPV-16 was the most prevalent infection HPV-positive in most oral cancers. Therefore, the prophylactic vaccine against HPV-16 could prevent HPV associated oral cancer if the vaccine were demonstrated to be capable of preventing oral HPV-16 infection. Thus, these findings have created new potential opportunities for the primary prevention of oral cancers.
Keywords: Human papillomavirus; Carcinogenesis; Squamous cell carcinomas; The upper aero-digestive tract.
Colomb Med. 2011; 42: 233-42
Infección por el VPH y la carcinogénesis en el tracto aero-digestivo superior
RESUMEN
Estudios moleculares y epidemiológicos han demostrado un papel etiológico del virus del papiloma humano (VPH) en un subconjunto de cánceres de las vías aerodigestivas superiores (VADS) en mujeres y hombres. Las infecciones orales por el VPH, adquiridas a través de sexo oral, parece ser el principal factor de riesgo para los cánceres orales asociados con este virus. La infección con el tipo de alto riesgo del VPH-16 es el genotipo más prevalente de los genotipos del VPH encontrados en la mayoría de los cánceres orales. Por lo tanto, la vacuna profiláctica en contra del VPH-16 podría ser útil para prevenir el cáncer oral si la vacuna es capaz de prevenir la infección oral por VPH. Así, estos resultados han creado nuevas oportunidades potenciales para la prevención primaria del cáncer oral.
Palabras clave:Virus del papiloma humano; Carcinogénesis; Carcinomas de células escamosas; Tracto aerodigestivo superior.
Colomb Med. 2011; 42: 233-42
Infectious agents are suspected to play causal roles in a variety of human malignancies. The public health impact of the oncogenic effects of these infections is considerable. Infection is estimated to be responsible for about 17.8% of all incident cases of cancer worldwide, accounting for 26.3% of all malignancies in economically developing countries and 7.7% in developed countries1.
The evaluation of causality for these infectious agents as human carcinogens is difficult, given their ubiquitous nature, the substantial length of time between infection, and the cancer event, the nature of cofactors, and the rarity of malignancy among those infected. Thus, a central problem for the epidemiologist is to define the natural history of infection and to identify those factors that are related to the development of cancer. Hence, informative biomarkers of the agent (such as viral load), of the host (such as abnormal antibody pattern), and of other oncogenic exposures (such as tobacco use) are required to understand viral-human interactions and develop interventions2.
Case-control studies have now recognized that human papillomavirus (HPV) infection in oral cavity is a strong risk factor for head and neck squamous cell carcinoma (HNSCC), and mostly for oropharyngeal cancer. Risk is increased for high-risk HPV-16 infection3. Therefore, HPV infection in the oral cavity has important health consequences, raising the need for further studies about these aspects to clarify the implications of an HPV diagnosis in oral cavity and HNSCC.
Human Papillomavirus (HPV). HPVs are nonenveloped icosahedral viruses with a diameter of 55 nm, belonging to the papillomaviridae family. This epitheliotropic virus has 72 capsomers enclosing an 8 kbp-long circular DNA genome. Although its DNA is double-stranded, only one strand contains open reading frames (ORF) that are transcribed. The viral DNA has eight ORFs and an upstream regulatory region, also called the long control region (LCR), which contains an origin of replication and cis-acting transcriptional regulatory elements4. Figure 1 shows the genome organization of HPV-16, the HPV type most strongly related to cervical cancer. The early region of the HPV genome contains six ORFs corresponding to E1, E2, E4, E5, E6, and E7 genes, which encode proteins necessary for viral replication and cell transformation. The late region codes for the two proteins of the viral capsid: L1, the major structural protein; and L2, the protein linking to encapsulated DNA4.
Classification of papillomaviruses. The L1 ORF is the most conserved gene in the papillomavirus (PV) genome and has, therefore, been used to identify new PV types over the past 15 years. A new PV isolate is recognized as a new PV type if the complete genome has been cloned and the DNA sequence of the L1 ORF differs by more than 10% from any known PV types. A difference between 2% and 10% homology defines a subtype, and less than 2%, a variant5.
HPV has more than 100 types, of which approximately 90 have already been characterized and assigned with numbers, and has five genuses: alpha-papillomavirus, beta-papillomavirus, gamma-papillomavirus, mu-papillomavirus, and nu-papillomavirus5. HPV is also subdivided into two major groups, cutaneous and mucosal, based on data from clinical manifestations6. Most mucosal HPV types exist in the genital area, which can be divided into high-risk and low-risk HPV types7. High-risk HPV types increase the risk of cervical cancer, which is almost always associated with HPV infection. To this date, approximately 20 HPV types have been identified as high-risk. Among them, HPV-16 and HPV-18 are considered to be associated with 70% of all cervical cancer. In contrast, low-risk HPV types, such as HPV-6 and HPV-11, cause genital warts but not cancer.
Furthermore, HPVs have intratype variants. However, information on variants is limited to certain HPV types. Yamada et al.8 showed five phylogenetic clusters with distinct geographic distributions, analyzing the sequences of E6, L1, and LCR of HPV-16 isolated from cervical samples collected worldwide. The AA (Asian American) variant was isolated mainly from Central and South America and Spain. The African variants 1 and 2 (Af1 and Af2) and Asian (As) variant were present mainly from Africa and Southeast Asia, respectively. In all regions other than Africa, European-350T (E-350T) prototype, as well as European-350G (E-350G) variant were detected.
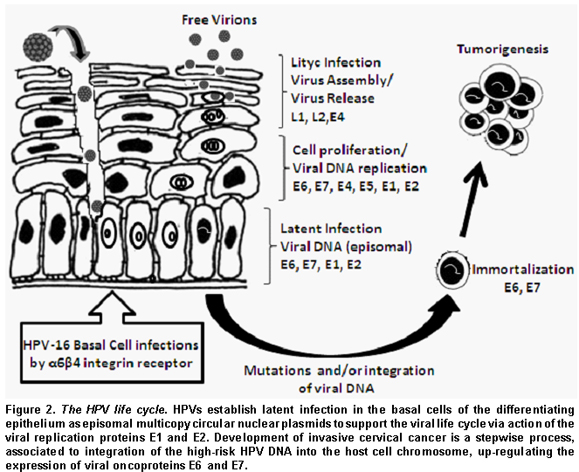
The HPV life cycle and its carcinogenesis. The life cycle of HPV is linked to the differentiation program of the infected host cell, epidermal or mucosal epithelial cell. Cells in the basal layer consist of stem cells, which persist indefinitely, and a much larger number of «transit amplifying cells», which arise from the stem cells and divide a finite number of times until they become diffe-rentiated, providing a reservoir of cells for subrabasal regions9.
HPV initially infects the basal layer of epithelia via minor abrasions. Viral entry into a cell is not clearly understood. It is suspected that the heparin sulfate mediates the initial attachment of virions to cells10 and that HPV enters a cell via interaction with certain receptors such as alfa-6 integrin for HPV-1611.
The first HPV genes to be expressed are E7 and E6 (Figure 2). The virus protein E7 promotes cell division by binding to pRb, a tumor suppressor protein that usually binds to and inactivates E2F, a transcription factor. E2F released from pRb causes transcription of genes involved in DNA replication and cell division. E6 virus protein binds to and inhibits p53 protein, which is active in repressing the cell cycle in the event of DNA damage, and in triggering apoptosis in the case of damage too severe to be repaired. E6 virus protein also activates cellular telomerase that synthesizes the telomere repeat sequences in eukaryotic cells and allows their immortal replication12. The transforming activities of high-risk HPV type represent a consequence of a viral replication strategy that is driven by the necessity to replicate the HPV genomes in suprabasal cells12. During the early phases of infection, the copy number of viral genome is between 50 and 100, and the viral genome exists as extrachromosomal plasmid or episomal form that replicates as the host cell chromosomes replicate. As the infected cells differentiate, the rest of the early viral genes, such as E1, E2, E4, and E5 genes, become switched on13. E1 and E2 proteins, a helicase and a transcription factor binding to LCR viral region, respectively, support viral DNA replication so that the infected stem cells can be maintained in the lesion for a long period. E4 viral protein is thought to be involved in activating the productive phase of the HPV life cycle. E5, another viral protein, is involved in transformation, enhancing the activity of EGF.
As infected daughter cells migrate to the upper layers of the epithelium, viral L1 and L2 late gene products, the major and minor viral capsid proteins, are produced to initiate the vegetative phase of the HPV life cycle, resulting in high-level amplification of the viral genome. At the upper layers of stratified squamous epithelia, viral DNA is packaged into capsids and virions produced are freed through normal desquamation processes, triggering little inflammation14. In addition, E6 and E7 proteins inactivate interferon regulatory factor15 so that HPV infection can remain persistent and asymptomatic.
Infection with high-risk HPV is associated with cervical dysplasia or cervical intraepithelial neoplasia (CIN). Long-term, persistent HPV infection in these lesions is thought to give rise to cervical cancers. CIN I (mild dysplasia) and CIN II (moderate dysplasia) lesions, in which the viral genomes replicate episomally, show relatively low levels of E6 and E7 gene expression, and are, in most cases, resolved spontaneously by an effective immune response. In contrast, CIN III (severe dysplasia, carcinoma in situ) and invasive cancer lesions, where viral DNA is integrated into the host genome in most cases, often display high-level expression of E6 and E7 genes16.
The integration of the viral genome into the host cell is a very rare event with a predilection for host chromosomal fragile sites17, but after it has happened carcinogenic transformation progresses rapidly. HPV integration into the host genome induces the increased E6 and E7 protein expressions since integration results in disruption of HPV E2 gene, which is a negative regulator of HPV E6 and E7 transcription. In addition, once integrated, the E6 and E7 mRNA gains a longer half-life by using host genome poly (A) signals. However, the ultimate development of cervical cancer is rarely accompanied by high expression of E6 and E7 proteins17.
High-risk HPV E6 and E7 oncoproteins can each independently induce genomic instability in normal human cells18. They cooperate to generate mitotic defects and aneuploidy through the induction of centrosome abnormalities in normal human epithelial cells, and the characteristic multipolar mitoses in cervical lesions are caused by centrosome abnormalities19. HPV oncoproteins expressing cells also exhibit centrosome-independent manifestations of genomic instability. These manifestations include anaphase bridges that may be caused by double-strand DNA breaks, as well as lagging chromosomal material20. To date, information on co-factors of HPV-related carcinogenesis in extra-genital organs is quite limited. Muñoz et al.21 proposed the following three groups of potential cofactors in cervical carcinogenesis:
SQUAMOUS CELL CARCINOMAS (SCC) OF THE UADT
Epidemiology. Oral cancer is the 11th most common cancer in the world in terms of number of cases, while cancer of the pharynx ranks as 20th. Worldwide, about 389,000 new cases occurred in 2000, two-thirds of which were in economically developing countries, and these cancers are responsible for some 200,000 deaths each year22. The male-female ratio of its incidence varies from 2 to 15, depending on the anatomical sub-site. An extremely high ratio is a characteristic of cancers of the tongue, floor of mouth, and pharyngeal. Cancers of the mouth and anterior two-thirds of the tongue are predominant in economically developing countries, whereas pharyngeal cancers are common in developed countries and in Central and Eastern Europe. In most countries, oral/pharyngeal cancer incidence and mortality rates have either been stable or increasing in the last four decades. Cancers of the esophagus are the sixth most frequent cancers worldwide. In 2000, the number of deaths due to esophageal cancer amounted to some 337,500 out of a total of 6.2 million cancer deaths worldwide. About 412,000 cases of cancer of the esophagus occur each year, of which over 80% are in economically developing countries23. The incidence of esophageal cancer shows a distinct geographical difference, which is more evident than for any other types of cancer. In certain regions in Asia, the incidence rates of ESCC are as high as 200 per every 100,000. Even within these high-risk areas, there are striking local variations in ESCC risk.
Genetic alterations. The genetic alterations observed in the cancers of the UADT include activation of protooncogenes such as cyclin D1, MYC, RAS, EGF receptor, HST-1, and HST-2, as well as inactivation of tumor suppressor genes (TSGs), such as those encoding p53 and p16INK4a25. Likewise, in cancer of the esophagus, the mutation of the p53 gene is detected in 35-70% of tumors, depending on geographic origin. Mutations in p53 have also been observed in dysplasia and in normal mucosa adjacent to cancer lesions, and considered an early event. The p16INK4a gene is another TSG that plays an important role in UADT development, and p16INK4a is often subject to hypermethylation of its promoter region, resulting in down-regulation of its expression26.
Etiology. Consumption of tobacco and alcohol, associated with low intake of fresh fruit, vegetables, and meat, is causally associated with SCCs of the UADT worldwide. However, the relative contribution of these risk factors varies from one geographic area to another27. Smoking is estimated to be responsible for about 41% of oral/pharyngeal cancers in men, and 15% in women worldwide. In more economically developed countries, it is estimated that 90% of ESCCs are attributable to tobacco and alcohol, with a multiplicative increase in risk when individuals are exposed to both factors. In addition, it has been reported that a genetic polymorphism of aldehyde dehydrogenase 2 (ALDH2), which plays a role in ethanol metabolism, is significantly associated with ESCC in the Japanese population28.
Other environmental risk factors include nitrosamines, deficiency of vitamins A and C, copper, and zinc, poor nutrition, and ingestion of pickled and preserved foods contaminated with fungi such as Aspergillus flavum, Geotrichum candidum, and Fusarium sp. Infectious agents, such as HPV and EBV, have also been suggested to be involved in the development of cancer of the UADT22.
HPV infection. The International Agency for Research on Cancer considers that there is convincing evidence that infection with HPV-16, -18, -31, -33, -35, -39, -45, -51, -52, -56, -58, -59 and -66 can lead to cervical cancer30. Regarding HPV-16, evidence supports its causal role in cancers of the vulva, vagina, penis, and anus. The association of HPV with cancers of the UADT is also suspected. The UADT consists of a complex mucosa-covered conduit for food and air that extends from the vermilion surface of the lips to the esophagus. Major malignancies observed in the UADT are cancers of the oral cavity, oropharynx, larynx, and esophagus. Among them, HPV-16 is strongly suspected to cause cancers of the oral cavity and oropharynx. Limited evidence is available for the association of HPV with cancers of the larynx and periungual skin, but insufficient evidence for the roles of HPVs in cancer of the esophagus30.
In our research group of the department of epidemiology and preventive medicine at Kagoshima University, Japan, we found HPV infections in 21 ESCC specimens (29%)31. Sequencing analysis of L1 region fragment identified HPV-16 genotype in six Colombian cases (13%) and in five Chilean cases (19%). Also, we found that a large proportion of ESCC specimens harbor HPV-16 genotype in the integrated form in a certain area with a high ESCC incidence in China32. In studies on the association of HPV with cancers of the oral cavity, oropharynx, and esophagus, using cancer specimens from Japan, Pakistan, and Colombia, we found HPV DNA in around half of the cases in SCCs of the oral cavity and oropharynx, and in a smaller proportion of ESCCs; the high-risk type HPV-16 was the most prevalent type; the viral load of HPV-16 in SCC of the tonsil was similar to that of cervical cancer and was higher than those of the other carcinomas (> to one copy per cell); and HPV-16 genomes detected in SCCs of the oral cavity, oropharynx, and esophagus were frequently integrated in the host genome (Data in publication process).
The most recent systematic review that included 5,046 SCC of head and neck cancers cases from 60 studies employing PCR-based methods showed that the presence of HPV DNA was 25.9%, being significantly higher in oropharyngeal SCC (35.6%; range 11%-100%) than in oral (23.5%; range 4%-80%) or laryngeal SCC (24.0%; range 0%-100%). HPV-16 accounted for a larger majority of HPV-positive oropharyngeal SCC (86.7%) than HPV-positive oral (68.2%) and laryngeal SCC (69.2%)33. In another meta-analysis of 4,680 samples from 94 reports published during the period between 1982 and 1997 showed that HPV was between 2 and 3 times more likely to be detected in precancerous oral mucosa and 4.7 times more likely to be detected in oral carcinoma than in normal mucosa34. Among the studies used in their meta-analysis, the largest-scale and best-designed study was the one by Maden et al.35 They examined 112 normal mucosa specimens and 118 oral carcinomas and detected HPV-16 in six cases of oral carcinomas, but only one sample of normal mucosa. On the other hand, HPV-6 was detected in 12 and 10 oral carcinomas and normal mucosa, respectively. A recent hospital-based case-control study of oropharyngeal cancer in the US detected HPV-16 DNA in 72% of 100 paraffin-embedded tumor specimens, and showed an association of oral HPV-16 infection with oropharyngeal cancer. The study also showed that 64% of patients with cancer were seropositive for the HPV-16 oncoproteins E6 or E7, or both36. HPV DNA in situ hybridization clearly showed its presence in the nuclei of cancer cells and not in surrounding normal cells. Another case-control study in the US found high-risk HPV, mainly HPV-16, more frequently in exfoliated oral cells from cancer patients than in those specimens from controls, suggesting an association of oral HPV infection with an increased risk of SCC in the head and neck37. In addition, elevated antibodies against L1 and/or E6/E7 were shown in an international study38.
A review study also showed that 15.2% of the 2,020 esophageal SCC cases tested by PCR until 2002 were HPV positive39. However, the role of HPV in esophageal carcinomas remains unclear and controversial. European prospective serologic studies that used stored serum specimens40 as well as a Chinese case-control study41 found a strong association between the risk of ESCCs and seropositivity to HPV-16. In contrast, other retrospective studies conducted in Europe42 and a large prospective serologic study in China43 found no significant association of HPV-16 or HPV-18 with SCCs or adenocarcinomas of the esophagus.
Recently, in the United States, Gillison44 showed that HPV is a causal factor for a distinct group of UADT cancers particularly in oropharyngeal cancers that occur more frequently in men than women, where oral sex appear to be the principal risk factor for HPV-associated oral cancers in adolescents45. Also, the tumor HPV status may be a strong and independent prognostic factor for survival among patients with oropharyngeal cancer46.
One important question is the route of HPV infection in the oral cavity, oropharynx, and esophagus tract. HPV is known to be sexually transmitted in the case of the anogenital organs4. However, limited available data suggest that HPV infection in oral cavity is possibly sexually acquired: a history of sexually transmitted diseases and number of oral sexual partners are associated with both oral HPV infection47 and HPV-positive oropharyngeal cancer36. Some data suggest that the presence and persistence of an oral high-risk HPV infection is associated with a persistent oral infection in a spouse,48 as well as in an increased risk for oral cancer among women with a history of cervical cancer and their husbands49. In addition, other several conceivable ways have been proposed for HPV infection in oral and pharyngeal cavities. These include intrapartum infection during the passage through the infected birth canal, transplacental infection in uterus prior to birth, and postnatal infection by contact. For instance, HPV can be transmitted from a mother to her newborn baby during vaginal delivery resulting in recurrent respiratory papillomatosis. In addition, HPV DNA has been detected in the foreskin of normal newborn and in a high percentage of neonates vaginally delivered by HPV-infected mothers, as well as in the amniotic fluid50.
Etiological role of HPV. Studies on prevalence of HPV infections in premalignant and malignant lesions of the oral cavity suggested the implication of HPV during the early stages of oral neoplasia and a role in malignant progression51. HPV-16 immortalized oral keratinocytes have demonstrated to accumulate progressive chromosomal aberrations52 and express high levels of differentiation; however, there was no tumorigenic activity in nude mice unless subjected to chronic exposure to the tobacco carcinogen benzo(a) pyrene53. Both benzo(a)pyrene stimulation and HPV-16 infection of cultured oral epithelial cells have been shown to confer anti-apoptotic characteristics, such as down-regulation of Fas and Bax, as well as over-expression of Bcl2 via p53 deregulation. Consequently, HPV alone is not sufficient to induce malignant transformation in several oral anatomic locations compared to oropharyngeal cancer and tonsillar cancer in particular. Futher studies are needed.
Methods for HPV detection. As HPVs cannot be cultured easily; usually, HPV detection and genotype assays are based on the detection of viral nucleic acids, mostly viral DNA. HPV-DNA is detected by target amplification methods and/or signal amplification methods. The most used target amplification-based method is polymerase chain reaction (PCR) using conserved sequences of the HPV genome, almost exclusively within the L1 open reading frame (ORF). In our first studies in UADT by using formalin-fixed and paraffin-embedded specimens31,32, we detected HPV-DNA by GP5+/GP6+ primer pair for PCR54 that amplified 150 base pair regions within L1, and the results were confirmed by southern blot analysis. Also, sequencing of L1 gene fragment amplified was used to identify HPV genotype. In our subsequent studies in UADT, using formalin-fixed and paraffin-embedded specimens, we used an ultrasensitive short-fragment PCR assay, the SPF10, which amplifies a 65-base-pair region within L155. The HPV types were determined by using the INNO-LiPA HPV genotyping v2 kit (Innogenetics NV, Belgium), based on the reverse hybridization principle. In brief, part of the L1 gene region of the HPV genome is amplified by using SPF10 primers tagged with a biotin at the 52 and denatured. Biotinylated amplicons are hybridized with specific oligonucleotide probes immobilized on the strip. In total, there were 25 genotypes (HPV-6, -11, -16, -18, -31 -33, -35, -39, -40, -42, -43, -44, -45, -51, -52, -53, -54, -56, -58, -59, -66, -68, -70, -73, and -74). Recently, real-time PCR assays have been used to determine the number of viral copies of HPV and to determine its integration status56. HPV-16 physical status is determined on the assumption that the E2 gene is disrupted in integrated viral genome and, therefore, the expected ratio of E2 to E6 copy numbers is zero. On the other hand, the episomal viral genome has equivalent copy numbers of the E2 and the E6 genes (an E2/E6 ratio is nearly equal to the unity) and mixed presence of integrated and episomal form HPV-16 have an E2/E6 ratio between 0 and 1. Other target amplification-based methods as reverse-transcriptase (RT-)PCR assays can be applied to detect HPV mRNAs in fresh-frozen specimens or samples in which RNA is well preserved (i.e., liquid-based cytology samples of cervical scrapings). Regarding signal amplification methods, these are based on an initial hybridization step of nucleic acids in the specimen with target-specific probes in liquid phase or in situ on cells or tissue slides, after which the signal (i.e., the hybridization event) is amplified and ultimately visualized with one of the various methodologies available. The liquid-phase signal amplification method Digene Hybrid Capture 2 (HC2) assay57 (Qiagen, Gaithersburg, MD, USA) is the first Food and Drug Administration (FDA) approved test that screens for the presence or absence of oncogenic HPV types. HC2 assay uses a mixture of RNA probes, representing 13 HPV genotypes (HPV-16, -18, -31, -33, -35, -39, -45, -51, -52, -56, -58, -59, and -68) to hybridize to HPV-DNA positive samples. DNA–RNA hybrids are subsequently captured in microplate wells coated with antibodies that specifically recognize DNA–RNA hybrids. For clinical validation strategy for candidate HPV tests, we need a clinical equivalence analysis of the candidate assay relative to a clinically validated reference HPV test (HC2) by non-inferiority testing using samples that originate from a population-based screening cohort.
HPV vaccine. Preventive HPV vaccines are based on empty virus-like particles (VLPs) assembled from recombinant HPV coat proteins. Two HPV vaccines are currently on the market: Gardasil and Cervarix. The vaccine works by making the individual immune to two key strains of the HPV (HPV-16 and -18). Together, the two strains are known to cause approximately 70% of all cervical cancer cases worldwide and some other genital cancers. Gardasil58 also protects against the two HPV types (HPV-6 and HPV-11) that cause 90% of genital warts. In the UK, the Joint Committee on Vaccination and Immunization recommended routine vaccination for 11 to 12-year olds, including the possibility of a catch-up campaign but only up to the age of 16. Similar recommendations on vaccination of young women against HPV to prevent cervical cancer were made by Public Health officials in Australia, Canada, Europe, and the United States59. It is suggested that vaccinating most teenage girls could save hundreds of lives a year, although, the benefits would not be seen until those receiving the vaccine enter middle age. Besides, Gardasil has been shown to prevent potential precursors to anal, vulvar, vaginal, and penile cancers. HPV vaccines are expected to protect against HPV induced cancers of these areas, as well as HPV-induced oral cancers. Also, laboratory research and clinical trials have focused on the development of therapeutic vaccines against HPV oncogenes, such as E6 and E7. It is hoped that immune responses against the two oncogenes might eradicate established tumors60.
CONCLUSIONS
The IARC estimated that 3% of oral cavity and 12% of oropharynx cancers worldwide are attributable to HPV1. Current generation HPV-16 and -18 L1 VLP vaccines hold potential promise for the prevention of a greater majority of HPV-positive cancers mainly HPV-associated cervical cancer in female adolescents. However, HPV vaccination in males may be a particularly important approach to prevent UADT cancer; most HPV-associated UADT cancers occur in men. So, with the knowledge accumulated in recent years on HPV-associated UADT cancers, there is reason to be optimistic that HPV vaccines may be protective against UADT HPV infection and, thus, be effective in preventing HPV-associated UADT cancers in both men and women. Hence, clinical trials to evaluate the efficacy of vaccines in protecting against HPV and its impact in HPV-associated UADT cancers are needed.
Conflict of interest. I hereby declare that the work submitted bears no potential conflicts of interest.
REFERENCES
1. Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006; 118: 3030-44.
2. Mueller NE, Birmann B, Parsonnet J, Schiffman M, Stuver S. Infectious agents. In: Schottenfeld D, Fraumeni JF Jr, (eds.). Cancer epidemiology and prevention. 3rd ed. New York: Oxford University Press; 2005.
3. D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papilloma virus and oropharyngeal cancer. N Engl J Med. 2007; 356: 1944-56.
4. zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002; 2: 342-50.
5. de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004; 324: 17-27.
6. Van Ranst M, Kaplan JB, Burk RD. Phylogenetic classification of human papillomaviruses: correlation with clinical manifestations. J Gen Virol. 1992; 73: 2653-60.
7. Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003; 348: 518-27.
8. Yamada T, Manos MM, Peto J, Greer CE, Munoz N, Bosch FX, et al. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J Virol. 1997; 71: 2463-72.
9. Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004; 68: 362-72.
10. Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, et al. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem. 1999; 274: 5810-22.
11. Yoon CS, Kim KD, Park SN, Cheong SW. alpha (6) Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem Biophys Res Commun. 2001; 283: 668-73.
12. Doorbar J. The papillomavirus life cycle. J Clin Virol. 2005; 32: S7-15.
13. Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004; 78: 11451-60.
14. Moscicki AB, Schiffman M, Kjaer S, Villa LL. Updating the natural history of HPV and anogenital cancer. Vaccine. 2006; 24: S42-51.
15. Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002; 179: 205-12.
16. Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004; 64: 3878-84.
17. Pett M, Coleman NJ. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J Pathol. 2007; 212: 356-67.
18. Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004; 109: 157-62.
19. Duensing S, Münger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003; 77: 12331-5.
20. Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002; 62: 7075-82.
21. Muñoz N, Castellsagué X, de González AB, Gissmann L. HPV in the etiology of human cancer. Vaccine. 2006; 24: S1-S10.
22. Stewart BW, Kleihues P (eds.). World Cancer Report. Lyon: IARC Press; 2003.
23. Schwartz JL. Biomarkers and molecular epidemiology and chemoprevention of oral carcinogenesis. Crit Rev Oral Biol Med. 2000; 11: 92-122.
24. Montesano R, Hollstein M, Hainaut P. Genetic alterations in esophageal cancer and their relevance to etiology and pathogenesis: a review. Int J Cancer. 1996; 69: 225-35.
25. Mandard AM, Hainaut P, Hollstein M. Genetic steps in the development of squamous cell carcinoma of the esophagus. Mutat Res. 2000; 462: 335-42.
26. Ruesga MT, Acha-Sagredo A, Rodríguez MJ, Aguirregaviria JI, Videgain J, Rodríguez C, et al. p16(INK4a) promoter hypermethylation in oral scrapings of oral squamous cell carcinoma risk patients. Cancer Lett. 2007; 250: 140-5.
27. Guha N, Boffetta P, Wünsch Filho V, Eluf Neto J, Shangina O, Zaridze D, et al. Oral health and risk of squamous cell carcinoma of the head and neck and esophagus: results of two multicentric case-control studies. Am J Epidemiol. 2007; 166: 1159-73.
28. Yokoyama A, Muramatsu T, Ohmori T, Yokoyama T, Okuyama K, Takahashi H, et al. Alcohol-related cancers and aldehyde dehydrogenase-2 in Japanese alcoholics. Carcinogenesis. 1998; 19: 1383-7.
29. Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F. WHO International Agency for Research on Cancer. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005; 6: 204.
30. Gillison ML, Shah KV. Role of mucosal human papillomavirus in nongenital cancers. J Natl Cancer Inst Monogr. 2003; 31: 57-65.
31. Castillo A, Aguayo F, Koriyama C, Torres M, Carrascal E, Corvalan A, et al. Human papillomavirus in esophageal squamous cell carcinoma in Colombia and Chile. World J Gastroenterol. 2006; 12: 6188-92.
32. Shuyama K, Castillo A, Aguayo F, Sun Q, Khan N, Koriyama C, et al. Human papillomavirus in high- and low-risk areas of esophageal squamous cell carcinoma in China. Br J Cancer. 2007; 96: 1554-9.
33. Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005;14:467-75.
34. Miller CS, Johnstone BM. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982-1997. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001; 91: 622-35.
35. Maden C, Beckmann AM, Thomas DB, McKnight B, Sherman KJ, Ashley RL, et al. Human papilloma viruses, herpes simplex viruses, and the risk of oral cancer in men. Am J Epidemiol. 1992; 135: 1093-102.
36. D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007; 356: 1944-56.
37. Smith EM, Ritchie JM, Summersgill KF, Hoffman HT, Wang DH, Haugen TH, Turek LP. Human papillomavirus in oral exfoliated cells and risk of head and neck cancer. J Natl Cancer Inst. 2004; 96: 449-55.
38. Herrero R, Castellsagué X, Pawlita M, Lissowska J, Kee F, Balaram P, et al. Human papillomavirus and oral cancer: the International Agency for Research on Cancer multicenter study. J Natl Cancer Inst. 2003; 95: 1772-83.
39. Syrjanen KJ. HPV infections and esophageal cancer. J Clin Pathol. 2002; 55: 721-8.
40. Bjorge T, Hakulinen T, Engeland A, Jellum E, Koskela P, Lehtinen M, et al. A prospective, seroepidemiological study of the role of human papillomavirus in esophageal cancer in Norway. Cancer Res. 1997; 57: 3989-92.
41. Han C, Qiao G, Hubbert NL, Li L, Sun C, Wang Y, et al. Serologic association between human papillomavirus type 16 infection and esophageal cancer in Shaanxi Province, China. J Natl Cancer Inst. 1996; 88: 1467-71.
42. Van Doornum GJ, Korse CM, Buning-Kager JC, Bonfrer JM, Horenblas S, Taal BG, et al. Reactivity to human papillomavirus type 16 L1 virus-like particles in sera from patients with genital cancer and patients with carcinomas at five different extragenital sites. Br J Cancer. 2003; 88: 1095-100.
44. Kamangar F, Qiao YL, Schiller JT, Dawsey SM, Fears T, Sun XD, et al. Human papillomavirus serology and the risk of esophageal and gastric cancers: Results from a cohort in a high-risk region in China. Int J Cancer. 2006; 119: 579-84.
44. Gillison ML. HPV and prognosis for patients with oropharynx cancer. Eur J Cancer. 2009; 45: 383-5.
45. Gillison ML. Human papillomavirus-related diseases: oropharynx cancers and potential implications for adolescent HPV vaccination. J Adolesc Health. 2008; 43: S52-60.
46. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010; 363: 24-35.
47. Scardina GA, Pisano T, Messina P. Oral and cervical lesions associated with human papillomavirus. Recenti Prog Med. 2009; 100: 261-6.
48. Rintala M, Grenman S, Puranen M, Syrjanen S. Natural history of oral papillomavirus infections in spouses: a prospective Finnish HPV Family Study. J Clin Virol. 2006; 35: 89-94.
49. Hemminki K, Dong C, Frisch M. Tonsillar and other upper aerodigestive tract cancers among cervical cancer patients and their husbands. Eur J Cancer Prev. 2000; 9: 433-7.
50. Puranen M, Yliskoski M, Saarikoski S, Syrjänen K, Syrjänen S. Vertical transmission of human papillomavirus from infected mothers to their newborn babies and persistence of the virus in childhood. Am J Obstet Gynecol. 1996; 174: 694-9.
51. Tsantoulis PK, Kastrinakis NG, Tourvas AD, Laskaris G, Gorgoulis VG. Advances in the biology of oral cancer. Oral Oncol. 2007; 43: 523-34.
52. Oda D, Bigler L, Mao EJ, Disteche CM. Chromosomal abnormalities in HPV-16-immortalized oral epithelial cells. Carcinogenesis. 1996; 17: 2003-8.
53. Park NH, Gujuluva CN, Baek JH, Cherrick HM, Shin KH, Min BM. Combined oral carcinogenicity of HPV-16 and benzo(a)pyrene: an in vitro multistep carcinogenesis model. Oncogene. 1995; 10: 2145-53.
54. De Roda Husman AM, Walboomers JM, van den Brule AJ, Meijer CJ, Snijders PJ. The use of general primers GP5 and GP6 elongated at their 3' ends with adjacent highly conserved sequences improves human papillomavirus detection by PCR. J Gen Virol. 1995; 76: 1057-62.
55. Kleter, B, Van Doorn LJ, Schrauwen L, Molijn A, Sastrowijoto S, Schegget J ter, et al. Development and clinical evaluation of a highly sensitive PCR-reverse hybridization line probe assay for detection and identification of anogenital human papillomavirus. J Clin Microbiol. 1999; 37: 2508-17.
56. Peitsaro P, Johansson B, Syrjanen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol. 2002; 40: 886-91.
57. Clavel C, Masure M, Putaud I, Thomas K, Bory JP, Gabriel R, et al. Hybrid capture II, a new sensitive test for human papillomavirus detection. Comparison with hybrid capture I and PCR results in cervical lesions. J Clin Pathol. 1998; 51: 737-40.
58. Garland SM, Smith JS. Human papillomavirus vaccines: current status and future prospects. Drugs. 2010; 70: 1079-98.
59. Allen JD, Coronado GD, Williams RS, Glenn B, Escoffery C, Fernandez M, et al. A systematic review of measures used in studies of human papillomavirus (HPV) vaccine acceptability. Vaccine. 2010; 28: 4027-37.
60. Lin K, Doolan K, Hung CF, Wu TC. Perspectives for preventive and therapeutic HPV vaccines. J Formos Med Assoc. 2010; 109: 4-24.